What is in this leaflet
This leaflet answers some common questions about BOSENTAN RBX.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using this medicine against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What BOSENTAN RBX is used for
BOSENTAN RBX is used for the treatment of high blood pressure in the blood vessels between the heart and the lungs. This condition is called pulmonary arterial hypertension.
This medicine acts to reduce abnormally high blood pressure by widening these blood vessels. It belongs to the class of medicines known as endothelin receptor antagonists.
Your doctor however, may prescribe BOSENTAN RBX for another purpose.
Ask your doctor if you have any questions why it has been prescribed for you.
This medicine is only available with a doctor’s prescription.
Before you take BOSENTAN RBX
When you must not take BOSENTAN RBX
Do not take this medicine if you are:
- Pregnant or intend to become pregnant. You must stop taking the medicine at least 3 months before trying to become pregnant. It is known that this medicine causes harm to the developing baby if you take it during pregnancy and in the three months before becoming pregnant.
- Breastfeeding: Tell your doctor immediately if you are breastfeeding. You are advised to stop breastfeeding if this medicine is prescribed for you because it is not known if this drug passes into the milk in women who are taking this medicine.
- Being treated with cyclosporine A (a medicine used after a transplant or to treat psoriasis)
- Being treated with glibenclamide (a medicine used for diabetes)
Do not take BOSENTAN RBX if you are allergic to it or any of the ingredients listed at the end of this leaflet.
Do not take BOSENTAN RBX if you have moderate to severe liver disorder.
Do not take it after the expiry date (EXP) printed on the pack. If you take it after the expiry date has passed, it may not work as well.
Do not take it if the packaging is torn or shows signs of tampering.
Before you start to take BOSENTAN RBX
BOSENTAN RBX may harm sperm. All men should use effective birth control while taking this medicine and for 3 months after they stop taking it.
If sexually active, you must use a hormonal and barrier method of contraception. This medicine may reduce the effectiveness of hormone contraceptives such as the pill and hormone patches, implants or injection. It is important to use other contraceptives, like condoms or an intrauterine device.
Your doctor will advise you about using reliable contraceptives before taking this medicine.
You must have a negative pregnancy test at the time of starting treatment if you are sexually active. Your doctor will need evidence that you are not pregnant.
Tell your doctor if:
- You are a woman of childbearing potential and not using reliable contraceptive methods. You must have a negative pregnancy test before beginning treatment. The test should be performed on the second day of a normal menstrual period or 11 days after the last unprotected sexual intercourse, whichever is later. Your doctor will advise you about using reliable contraception before taking or whilst taking this medicine. Hormonal contraception on its own is not a reliable option because this medicine may make this method ineffective in preventing pregnancy. Hormonal contraceptives include ones you take orally (the pill), patches you put on your skin, ones that are injected and implants. You should always use additional methods of contraception such as condoms and IUDs and not rely just on hormonal contraception. You should have a pregnancy test every month while you are taking this medicine. You must stop taking this medicine for at least 3 months prior to becoming pregnant.
- You are breastfeeding or planning to breastfeed. It is not known whether BOSENTAN RBX passes into breast milk.
- You have allergies to:
- any other medicines
- any other substances such as foods, preservatives or dyes.
- You have or have had any medical conditions, especially the following:
- pulmonary arterial hypertension
- anaemia
- hypotension
- liver or renal disorders
- heart failure
- HIV infection
If you have not told your doctor about any of the above, tell them before you use BOSENTAN RBX.
While taking BOSENTAN RBX
Do not become pregnant while taking this medicine.
You must have a pregnancy test every month while you are taking this medicine. Your doctor will need evidence that you are not pregnant before prescribing this medicine again.
Interactions with other medicines
Tell your doctor if you are taking any other medicines including any that you buy without a prescription from your pharmacy, supermarket or health food shop. Some medicines may interfere with BOSENTAN RBX. These include:
- hormonal contraceptives (oral, injectable, transdermal and implantable)
- simvastatin, medicines for lowering blood fats
- medicines for diabetes such as glibenclamide
- medicines for fungal infections such as ketaconazole, itraconazole, fluconazole and voriconazole
- medicines for bacterial infections such as rifampicin
- medicine to prevent organ transplantation rejection such as Cyclosporine A, tacrolimus and sirolimus
- medicines for rheumatoid arthritis or psoriasis/dermatitis
- lopinavir+ritonavir or other ritonavir-boosted protease inhibitors (used to treat HIV);
- digoxin (used to treat heart rhythm disorders)
- sildenafil (used to treat erectile dysfunction and/or also pulmonary hypertension
- nevirapine (an HIV medicine)
These medicines may be affected by BOSENTAN RBX or may affect how well it works. You may need to use different amounts of your medicines, or take different medicines. Your doctor will advise you.
Your doctor or pharmacist has more information on medicines to be careful with or to avoid while taking BOSENTAN RBX.
How to take BOSENTAN RBX
How much to take
Always take this medicine exactly as your doctor has instructed you. You should check with your doctor or pharmacist if you are unsure.
The usual dose is one tablet, twice daily. For the first 4 weeks you will take a 62.5 mg tablet twice daily.
Depending on how you respond to the medicine, your doctor may increase the dosage after four weeks to a 125 mg tablet twice daily.
If you do not think the medicine is working or you think it is working too well, talk to your doctor. Your doctor may need to change the dose you are taking.
How to take this medicine
BOSENTAN RBX is taken by mouth, in the morning and evening, and can be taken with or without food.
How long to take it
Do not stop taking this medicine unless your doctor tells you to. Stopping your treatment may lead to a worsening of your symptoms. Your doctor may tell you to reduce the dose over a few days before stopping completely.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take the next dose when you are meant to.
Do not take a double dose to make up for the dose you have missed. This may increase the chance of getting an unwanted side effect.
If there is still a long time to go before your next dose, take it as soon as you remember and then go back to taking it as you would normally.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) or go to Accident and Emergency at your nearest hospital if you think you or anyone else may have taken too much BOSENTAN RBX.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using BOSENTAN RBX
Things you must do
It is very important that you have a liver function blood test before you start treatment and every month after that. BOSENTAN RBX can cause liver damage if it is not found early. Because this side effect may not cause symptoms at first, only a blood test can show that you have early liver damage. Regular blood tests let your doctor change or stop your therapy before there is permanent damage.
You should have a blood test for anaemia after 1 and 3 months, and then every 3 months for the rest of your treatment.
You need to have pregnancy tests monthly if you are a female of childbearing age
Things you must not do
Do not give the tablets to anyone else even if they have the same symptoms as you.
Things to be careful of
If you feel dizzy whilst taking this medicine, do not drive or operate any tools or machinery.
Side Effects
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- shortness of breath or difficulty in breathing
- nausea
- vomiting
- fever
- unusual tiredness
- stomach pain
- yellowing of your skin or the whites of your eyes (jaundice)
These may be serious side effects of BOSENTAN RBX. You may need urgent medical attention.
Serious side effects are uncommon.
Other side effects of this medicine include:
- Headache
- Inflamed throat and irritated nose passages
- Flushing (hot flashes)
- Ankle and leg swelling
- Low blood pressure
- Blood disorders
- Fast heart beat
- Tiredness
- Itching, rash, skin inflammation, skin redness
- Diarrhoea
See your doctor if any of these worry you.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed above may occur in some consumers.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using BOSENTAN RBX
Storage
Store this medicine in the original bottle, in a cool, dry place where the temperature stays below 25°C.
Keep out of reach of children.
Product Description
What it looks like
BOSENTAN RBX (bosentan) 62.5 mg film-coated tablets are light peach to peach coloured, round shaped, biconvex and debossed with ‘62.5’ on one side and plain on the other side.
BOSENTAN RBX (bosentan) 125 mg film-coated tablets are light peach to peach coloured, oval shaped, biconvex and debossed with ‘125’ on one side and plain on the other side.
Pack size
Supplied in PVC/PE/PVdC/Al blister packs and HDPE bottles containing 56 and 60 tablets.
Not all pack sizes and presentations are marketed in Australia.
Ingredients
Active ingredient:
BOSENTAN RBX 62.5 mg: 62.5 mg bosentan per tablet.
BOSENTAN RBX 125 mg: 125 mg bosentan per tablet.
Inactive ingredients:
maize starch, pregelatinised maize starch, sodium starch glycollate type A, povidone, glycerol dibehenate, magnesium stearate, hypromellose, glycerol triacetate, purified talc, titanium dioxide, iron oxide yellow, iron oxide red and ethylcellulose.
Sponsor
Sun Pharma ANZ Pty Ltd
Macquarie Park NSW 2113
Australia
[email protected]
Tel: 1800 726 229
This leaflet was prepared in March 2024
Australian Register Number:
BOSENTAN RBX 62.5 mg blister pack– 257757
BOSENTAN RBX 62.5 mg bottle– 257758
BOSENTAN RBX 125 mg blister pack– 257753
BOSENTAN RBX 125 mg bottle– 257756
Published by MIMS July 2024

 Additional adverse events occurring in the subset of patients treated for PAH, in at least 3% of patients on bosentan and more frequently than on placebo are presented in Table 3.
Additional adverse events occurring in the subset of patients treated for PAH, in at least 3% of patients on bosentan and more frequently than on placebo are presented in Table 3. Treatment with bosentan has been associated with dose-dependent elevations in liver aminotransferases and decreases in haemoglobin concentration (see Section 4.4 Special Warnings and Precautions for Use).
Treatment with bosentan has been associated with dose-dependent elevations in liver aminotransferases and decreases in haemoglobin concentration (see Section 4.4 Special Warnings and Precautions for Use).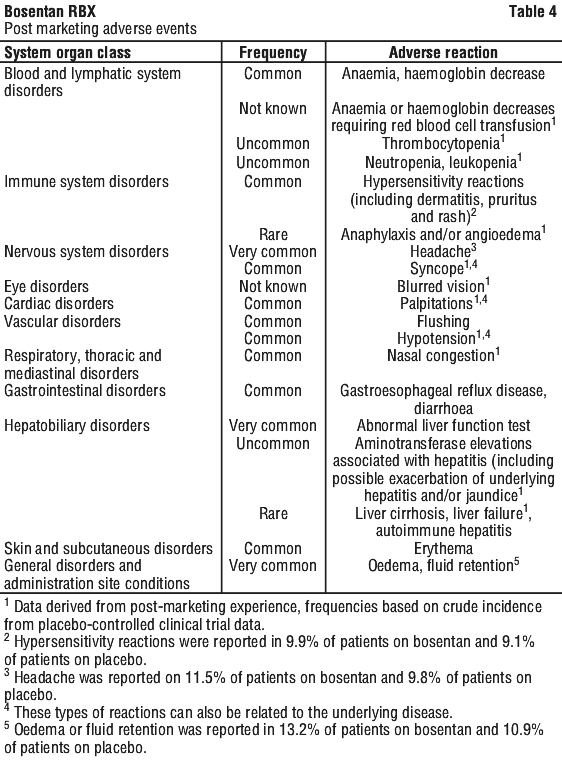 In the post-marketing period rare cases of unexplained hepatic cirrhosis were reported after prolonged therapy with bosentan in patients with multiple co-morbidities and drug therapies. There have also been rare reports of liver failure. These cases reinforce the importance of strict adherence to the monthly schedule for monitoring of liver function for the duration of treatment with bosentan.
In the post-marketing period rare cases of unexplained hepatic cirrhosis were reported after prolonged therapy with bosentan in patients with multiple co-morbidities and drug therapies. There have also been rare reports of liver failure. These cases reinforce the importance of strict adherence to the monthly schedule for monitoring of liver function for the duration of treatment with bosentan. In both trials, treatment with bosentan resulted in a significant increase in exercise capacity. The improvement in walk distance was apparent after 1 month of treatment (with 62.5 mg twice daily) and fully developed by about 2 months of treatment (see Figure 1). It was maintained for up to 7 months of double-blind treatment. The placebo-subtracted mean increase in walking distance was somewhat greater with 250 mg twice daily (54 m) than with 125 mg twice daily (35 m). However, the higher dose is not recommended because of the potential for increased liver injury (see Section 4.2 Dose and Method of Administration).
In both trials, treatment with bosentan resulted in a significant increase in exercise capacity. The improvement in walk distance was apparent after 1 month of treatment (with 62.5 mg twice daily) and fully developed by about 2 months of treatment (see Figure 1). It was maintained for up to 7 months of double-blind treatment. The placebo-subtracted mean increase in walking distance was somewhat greater with 250 mg twice daily (54 m) than with 125 mg twice daily (35 m). However, the higher dose is not recommended because of the potential for increased liver injury (see Section 4.2 Dose and Method of Administration). Change from baseline in 6-minute walking distance from start of therapy to week 16 in the placebo and combined bosentan (125 mg and 250 mg twice daily) groups. Values are expressed as mean ± standard error of the mean.
Change from baseline in 6-minute walking distance from start of therapy to week 16 in the placebo and combined bosentan (125 mg and 250 mg twice daily) groups. Values are expressed as mean ± standard error of the mean.
 There are limited data available on the minimum effective dose, dose response, and the clinically useful dose-range for bosentan.
There are limited data available on the minimum effective dose, dose response, and the clinically useful dose-range for bosentan. Patients with WHO functional class II PAH, on average, have only moderately impaired 6MWT and may therefore have a limited response range for improvement in this parameter. This could partly explain the lack of statistical significance for the 6MWT endpoint. However, there was a clear association between an absence of deterioration from baseline in 6MWT and stable WHO functional class in the EARLY population. No patient in the bosentan group who at least maintained baseline 6MWT had deterioration in functional class.
Patients with WHO functional class II PAH, on average, have only moderately impaired 6MWT and may therefore have a limited response range for improvement in this parameter. This could partly explain the lack of statistical significance for the 6MWT endpoint. However, there was a clear association between an absence of deterioration from baseline in 6MWT and stable WHO functional class in the EARLY population. No patient in the bosentan group who at least maintained baseline 6MWT had deterioration in functional class. The EARLY study was designed and powered to evaluate the efficacy of bosentan in the patient population as a whole and was not powered to show statistical significance for each aetiological sub-group. No criteria to determine aetiological subgroup numbers or specific claims of a beneficial effect by subgroup were pre-specified. Subgroup analyses of treatment effects according to PAH aetiology were performed in the EARLY trial with the objective being to support absence of heterogeneity in treatment response between subgroups. For both co-primary endpoints the confidence intervals for treatment effects were overlapping between the major aetiological subgroups. The inclusion criteria for EARLY permitted recruitment of any patient with PAH determined to be idiopathic/familial or secondary to congenital heart defect, or connective tissue disease and other predefined aetiologies. For patients with congenital heart disease the defect had to be isolated and restrictive with no reverse shunt (atrial septum defect (ASD) < 2 cm, ventricular septum defect (VSD) < 1 cm or patent ductus arteriosus (PDA)). The population eventually enrolled reasonably reflected the relative incidences of PAH aetiologies seen in the real world setting. Consequently there was more data available for analysis in the most prevalent aetiological subgroups of idiopathic/familial PAH compared with the other subgroups.
The EARLY study was designed and powered to evaluate the efficacy of bosentan in the patient population as a whole and was not powered to show statistical significance for each aetiological sub-group. No criteria to determine aetiological subgroup numbers or specific claims of a beneficial effect by subgroup were pre-specified. Subgroup analyses of treatment effects according to PAH aetiology were performed in the EARLY trial with the objective being to support absence of heterogeneity in treatment response between subgroups. For both co-primary endpoints the confidence intervals for treatment effects were overlapping between the major aetiological subgroups. The inclusion criteria for EARLY permitted recruitment of any patient with PAH determined to be idiopathic/familial or secondary to congenital heart defect, or connective tissue disease and other predefined aetiologies. For patients with congenital heart disease the defect had to be isolated and restrictive with no reverse shunt (atrial septum defect (ASD) < 2 cm, ventricular septum defect (VSD) < 1 cm or patent ductus arteriosus (PDA)). The population eventually enrolled reasonably reflected the relative incidences of PAH aetiologies seen in the real world setting. Consequently there was more data available for analysis in the most prevalent aetiological subgroups of idiopathic/familial PAH compared with the other subgroups.
