What is in this leaflet
This leaflet answers some common questions about Brenzys.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Brenzys against the benefits this medicine is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Brenzys is used for
Brenzys is used in adults for the treatment of:
- rheumatoid arthritis
- ankylosing spondylitis
- non-radiographic axial spondyloarthritis (nr-AxSpA)
- psoriatic arthritis
- plaque psoriasis.
Tumour necrosis factor (TNF) is a naturally occurring chemical messenger in your bloodstream. TNF plays a role in causing pain and swelling in the joints. Brenzys is a biotechnology-derived protein that works by binding to TNF and preventing it from acting. This reduces the pain and swelling of rheumatoid arthritis and psoriasis, helps to treat the skin lesions of psoriasis and psoriatic arthritis, and improves the condition of patients with ankylosing spondylitis and nr-AxSpA.
Your doctor may have prescribed Brenzys for another reason. Ask your doctor if you have any questions about why Brenzys has been prescribed for you.
This medicine is available only with a doctor's prescription.
Brenzys is not addictive.
Brenzys is not indicated for use in children less than 18 years of age. Other etanercept products are available for use in children.
Before you use Brenzys
When you must not use it
Do not use Brenzys if:
- You have an allergy to Brenzys or any of the ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction may include wheezing, shortness of breath, difficulty breathing, or a tight feeling in your chest, swelling of the face, lips, tongue or other parts of the body, rash, itching, hives or flushed red skin, dizziness or light-headedness.
- You have, or are at risk of developing, sepsis (blood poisoning), or long-term or localised infection (for example, leg ulcer).
Sepsis is a serious infection causing fever, headache, joint aches and pains, sore or weak muscles, and increased heart rate. Brenzys can affect your body's ability to fight a serious infection.
If you are not sure whether you have a serious infection, check with your doctor.
- You are currently taking anakinra or other similar medicines known as Interleukin-1 antagonists, or other biological medicines such as anti-TNF drugs, tocilizumab or abatacept.
- The packaging is torn or shows signs of tampering.
- The expiry date (EXP) printed on the pack has passed.
If you use Brenzys after the expiry date has passed, it may have no effect at all, or worse, have an entirely unexpected effect.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you use it
Tell your doctor if:
- You have any allergies to:
- Any other medicines
- Any other substances, such as foods, preservatives or dyes.
- You are pregnant or intend to become pregnant.
The effects of Brenzys in pregnant women are not well understood, therefore Brenzys should only be used during pregnancy if clearly needed. If you become pregnant while using Brenzys, contact your doctor. Your doctor will help you to decide whether the benefits of treatment outweigh the potential risk to your baby.
- You are breast-feeding or plan to breast-feed.
Brenzys passes through breast milk. Therefore, if you are breast-feeding, you should discuss with your doctor whether to stop breast-feeding or stop using Brenzys.
- You have or have had any other medical conditions, especially the following:
- Serious infection including sepsis, tuberculosis or a history of recurring infections
- Low resistance to disease
- Diabetes
- Liver problems or hepatitis B or hepatitis C, viruses that affect the liver
- Heart failure
- Blood disorders
- Cancer
- Are about to have major surgery
- Nerve disorders including multiple sclerosis or optic neuritis (inflammation of the nerves of the eyes)
- Seizures
- Chicken pox or have been recently exposed to chickenpox.
Live vaccines, such as oral polio vaccine, should not be given while receiving Brenzys.
Ensure that children are up to date with all vaccinations (including chicken pox) before they start treatment with etanercept.
If you have not told your doctor about any of the above, tell them before you start using Brenzys.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may affect the way others work. Your doctor or pharmacist will be able to tell you what to do when using Brenzys with other medicines. Brenzys may interact with other medicines such as:
- Abatacept, sulfasalazine or Interleukin-1 antagonists such as anakinra. These medicines are used to treat rheumatoid arthritis and other inflammatory diseases
- Cyclophosphamide, a medicine used to treat cancer or prevent transplant rejection
- Some vaccines
- Digoxin, a medicine used to improve the strength and efficiency of the heart, or to control the rate and rhythm of the heartbeat
- Warfarin, a medicine used to thin the blood and prevent blood clots
- Medicines used to treat diabetes.
How to use Brenzys
After allowing the Brenzys syringe or Auto-injector to reach room temperature (approximately 30 minutes), immediate use is recommended.
Each syringe or Auto-injector of Brenzys is for single use only, in one patient only. Discard any residue.
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you are injecting Brenzys yourself, you must follow the detailed instructions provided in the leaflet inside the pack.
Brenzys is injected under the skin. When using the syringe or Auto-injector, it is important that you do not pull back on the plunger. Brenzys can be injected by your doctor, nurse, carer or by yourself.
When you have finished injecting Brenzys, discard the syringe or Auto-injector into a sharps container.
If you do not understand the instructions for injecting Brenzys found in the carton, ask your doctor or pharmacist for help.
To help you remember, use a diary to write in the days of the week you should have an Brenzys injection.
How much to use
Your doctor will tell you how to inject Brenzys. A different site should be used for each new injection. Each new injection should be given at least 3cm from an old site.
Adults
The recommended dose for adults is 50 mg per week, injected under the skin. Your doctor may determine a different frequency at which to inject Brenzys. If you are being treated for psoriasis, your doctor may prescribe a higher dose of Brenzys when you first begin your treatment. If you are being treated for nr-AxSpA and Brenzys has no effect on your condition within 12 weeks, your doctor may tell you to stop using this medicine.
Children
Brenzys is not indicated for use in children less than 18 years of age. Other etanercept products are available for use in children.
How long to take it
You should continue to inject Brenzys for as long as your doctor recommends.
Never inject more than the dose recommended by your doctor.
If you feel that the effect of Brenzys is too strong or too weak, talk to your doctor or pharmacist.
If you forget to take it
If you forget to give yourself an injection, you should inject the next dose as soon as you remember if it is within 48 hours since the scheduled dose time. If it is more than 48 hours since the last dose was due, wait for the next scheduled dose.
Do not use a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to use your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (tel 13 11 26) for advice or go to Accident and Emergency at your nearest hospital, if you think that you or anyone else may have used too much Brenzys. Do this even if there are no signs of discomfort or poisoning. Always take the labelled medicine carton with you, even if it is empty.
You may need urgent medical attention. There is very limited data on overdose with Brenzys. Ask your doctor if you have any concerns.
While you are using Brenzys
Things you must do
Tell any other doctors, dentists and pharmacists who are treating you that you are using Brenzys.
Tell your doctor if Brenzys is not making your condition better.
If you have or develop any serious infection while using Brenzys, do not inject any more Brenzys and contact your doctor immediately.
Seek medical advice immediately if you have any symptoms such as persistent fever, sore throat, bruising, bleeding or paleness. These symptoms may point to the existence of a potentially life threatening blood disorder, which may require you to stop taking Brenzys.
Be careful driving or operating dangerous machinery until you know how it affects you.
It is not known whether Brenzys causes dizziness or drowsiness.
Things you must not do
Do not shake the Brenzys syringe or Auto-injector.
Do not give Brenzys to anyone else even if they have the same condition as you.
Do not use Brenzys to treat any other complaints unless your doctor tells you to.
Do not stop using Brenzys, or lower the dosage, without checking with your doctor.
Do not stop using Brenzys because you are feeling better, unless your doctor advises you to. Your condition may flare up if you reduce the dose or stop using Brenzys.
Side effects
Check with your doctor as soon as possible if you have any problems while taking Brenzys, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
Like other medicines, Brenzys can cause some side effects. If they occur, most are likely to be minor and temporary. However, some may be serious and need medical attention. The most common side effects with Brenzys are injection site reactions, infections, allergic reactions, itching, rash and fever. Please see below for more detail.
Ask your doctor or pharmacist to answer any questions you may have.
Injection site reactions
The most common side effect is a mild reaction in the area where Brenzys was injected, including:
- Itching
- Bruising
- Redness
- Bleeding, swelling, pain or hardness around the injection site.
These reactions generally do not occur as often after the first month of treatment.
If you are concerned about injection site reactions, contact your doctor or pharmacist. When injecting Brenzys, some people have developed a reaction to an injection site used before.
Infections
Infections, including colds and sinus infections, are very common.
Serious infections may occur including tuberculosis and blood poisoning.
Tell your doctor immediately if you notice any of the following:
- Signs of an infection such as fever, chills, mouth ulcers or sore throat
- Signs of lung disease such as breathlessness during exercise or a dry cough
- Signs of soft tissue infections such as bumps or sores that do not heal, are swollen, red or have pus
- Signs of nervous system disorders such as seizures, numbness or tingling throughout your body, weakness in your arms and/or legs and dizziness, or problems with your eye sight
- Inflammation of the spinal cord
- Inflammation of the inner eye
- Inflammation of blood vessels in the skin or lymph glands
- Signs of inflammatory bowel disease such as diarrhoea or mucus or blood in your stools, stomach cramps, fever or weight loss
- Signs of an allergic reaction such as swelling of the face, lips or tongue which may cause difficulty in swallowing or breathing, or hives
- Signs of lupus or lupus-like syndrome, such as weight changes, persistent rash, fever, joint or muscle pain, or fatigue
- Other signs of immune system disorders such as skin rash, blisters and ulcers on the skin, in the mouth or airways
- Signs of a blood disorder such as tiredness, headaches, shortness of breath when exercising, dizziness, paleness, nose bleeds, unusual bleeding or bruising or more frequent infections
- Signs of heart failure such as shortness of breath, persistent cough, difficulty exercising, fast or irregular heartbeat, swelling in the legs or feet, tiredness and weakness.
These may be very serious side effects. You may need urgent medical attention or hospitalisation. These side effects are rare.
Other side effects not listed above may also occur in some patients. Tell your doctor if you notice anything else that is making you unwell.
There have been reports of some types of cancer developing in patients using Brenzys and other TNF blocking medicines. These include skin cancers, cancers that affect the lymph system called lymphoma and Kaposi’s sarcoma (which also affects the organs, skin, mouth, nose or throat), or affect the blood system called leukaemia. The role of Brenzys in the development of cancer is not known.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using Brenzys
Storage
Keep Brenzys where children cannot reach it.
Keep Brenzys in a refrigerator where the temperature stays between 2°C and 8°C. Do not freeze. Do not store Brenzys or any other medicine in the bathroom or near a sink. Do not leave it in the car or on window sills. Brenzys syringes and Auto-injectors should be stored in their cartons to protect them from light. Heat and dampness can destroy some medicines.
If it is not possible to store Brenzys in the refrigerator, it may be stored out of the refrigerator (below 25°C) for up to 4 weeks (e.g. when travelling). If you have stored Brenzys at room temperature for any period of time (even if returned to the refrigerator) you must use it within 4 weeks from the time you first took it out of the refrigerator, or else you must discard it. Do not use Brenzys if it has been exposed to high temperatures, or has been out of the refrigerator for more than 4 weeks.
Disposal
Ask your pharmacist how to dispose of medicines no longer required.
Product description
What it looks like
Pre-filled Syringe (Solution for injection):
Brenzys Pre-filled Syringe is supplied in a carton containing four single-use pre-filled glass syringes containing Brenzys solution. Each syringe contains 50 mg of etanercept in 1 mL of Brenzys solution.
Auto-injector (Solution for injection)
The Brenzys Auto-injector is supplied in a carton containing either one or four single-use pre-filled glass syringes, each housed in a plastic Auto-injector. Each syringe contains 50 mg of etanercept in 1 mL of Brenzys solution.
Ingredients
Solution for injection:
Brenzys solution for injection contains 50 mg of the active ingredient etanercept (rch). The Brenzys solution also contains sucrose, sodium chloride, sodium phosphate monobasic monohydrate, sodium phosphate dibasic (heptahydrate) and water.
Sponsor
Brenzys is supplied by:
Sponsor:
SAMSUNG BIOEPIS AU PTY LTD
Level 16, 201 Elizabeth Street,
Sydney NSW 2000, Australia
Distributor:
Organon Pharma Pty Limited Building A, 26 Talavera Road, Macquarie Park, NSW 2113, Australia
Australian Registration Number:
50 mg pre-filled syringe solution for injection: AUST R 245252
50 mg Auto-injector solution for injection: AUST R 245253
Date of preparation
This leaflet was prepared in March 2022.
® Registered Trademark
Instructions for Use
The following instructions are for preparing and giving a dose of BRENZYS® using a single-use pre-filled syringe.
YOUR BRENZYS® PRE-FILLED SYRINGE

STEP 1: GATHER SUPPLIES
- Place your syringe and unopened alcohol swabs on a clean, dry surface.
- Remember to wash your hands!
- Don’t remove the cap just yet!

Step 2: WAIT 30 MINUTES
- Wait approximately 30 minutes for your syringe to warm-up to room temperature, which helps reduce your pain during injection.
- Don’t remove the cap just yet!

Step 3: INSPECT MEDICINE & DATE
- Always make sure your medicine hasn’t expired.
- The medicine should be clear or slightly opalescent, colourless or pale yellow, and may contain small white or almost transparent particles of protein.
- You may see an air bubble, and that’s okay.
- Don’t remove the cap just yet!

Step 4: CHOOSE SITE & CLEAN SKIN
- Choose an injection site on your body.
- Your abdomen or thighs are best.
- Wipe your skin at the injection site with an alcohol swab.
- Avoid skin that’s sore, bruised, scarred, scaly or has red patches.

Step 5: REMOVE SYRINGE CAP
- Carefully remove the needle cap.

Step 6: PINCH SKIN & INSERT NEEDLE
- Gently pinch your skin, and carefully insert the needle.

Step 7: PUSH PLUNGER ALL THE WAY
- Hold the syringe steady and press the plunger all the way down.

Step 8: REMOVE SYRINGE & DISPOSE
- Pull the syringe away from your skin and dispose of it in a sharps container.
- Don’t recap or reuse your needle.

Instructions for Use
The following instructions are for preparing and giving a dose of BRENZYS® using a single-use auto-injector.
YOUR BRENZYS® AUTO-INJECTOR

Step 1: GATHER SUPPLIES
- Place your auto-injector and unopened alcohol swabs on a clean, dry surface.
- Remember to wash your hands!
- Don’t remove the cap just yet!

Step 2: WAIT 30 MINUTES
- Wait approximately 30 minutes for your auto-injector to warm-up to room temperature, which helps reduce your pain during injection.
- Don’t remove the cap just yet!

Step 3: INSPECT MEDICINE & DATE
- Always make sure your medicine hasn’t expired.
- The medicine should be clear or slightly opalescent, colourless or pale yellow, and may contain small white or almost transparent particles of protein.
- You may see an air bubble, and that’s okay.
- Don’t remove the cap just yet!

Step 4: CHOOSE SITE & CLEAN SKIN
- Choose an injection site on your body.
- Your abdomen or thighs are best.
- Wipe your skin at the injection site with an alcohol swab.
- Avoid skin that’s sore, bruised, scarred, scaly or has red patches.

Step 5: REMOVE THE BLUE NEEDLE CAP
- Carefully remove the blue needle cap with a metal center from the auto-injector.

Step 6: PLACE GRAY NEEDLE SHIELD, PRESS DOWN, AND HOLD 15 SECONDS
- Place the gray needle shield straight on your skin, and push the entire auto-injector down firmly to start the injection.
- When you push down, the injection starts.
- You may hear a click.

Step 7: AFTER 15 SECONDS, REMOVE AUTO-INJECTOR
- Hold the auto-injector against your skin.
- After 15 seconds, remove the auto-injector from the injection site.
- The medication window will change to yellow once the injection is completed.
- You may hear a second click.

Step 8: CONFIRM COMPLETION & DISPOSE AUTO-INJECTOR
- Confirm that the medication window is yellow.
- Discard your auto-injector in a sharps container.
- As per illustration a small grey band may still be visible.
- If the window isn’t yellow, you may not have received your full dose.

Published by MIMS May 2022


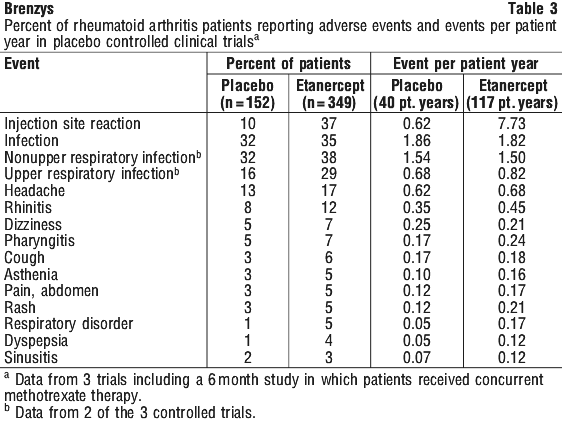 Based on the results of clinical studies in rheumatoid arthritis, normally no special laboratory evaluations are necessary in addition to careful medical management and supervision of patients.
Based on the results of clinical studies in rheumatoid arthritis, normally no special laboratory evaluations are necessary in addition to careful medical management and supervision of patients.
 The safety profiles during the open-label, extension period (after Week 52 up to Week 100) were also comparable between the treatment groups. A total of 60 (47.6%) patients from the Brenzys/Brenzys treatment group and 58 (48.7%) patients from the Enbrel/Brenzys treatment group were newly reported to have TEAEs. In 17 (13.5%) patients of the Brenzys/Brenzys treatment group and 16 (13.4%) patients of the Enbrel/Brenzys treatment group, these newly reported TEAEs were considered to be related to the treatment. No new cases of injection site reactions were reported in either treatment group.
The safety profiles during the open-label, extension period (after Week 52 up to Week 100) were also comparable between the treatment groups. A total of 60 (47.6%) patients from the Brenzys/Brenzys treatment group and 58 (48.7%) patients from the Enbrel/Brenzys treatment group were newly reported to have TEAEs. In 17 (13.5%) patients of the Brenzys/Brenzys treatment group and 16 (13.4%) patients of the Enbrel/Brenzys treatment group, these newly reported TEAEs were considered to be related to the treatment. No new cases of injection site reactions were reported in either treatment group. Two (1.7%) cases of oral candidiasis and 2 (1.7%) cases of cardiac failure were reported for patients who switched from Enbrel to Brenzys treatment.
Two (1.7%) cases of oral candidiasis and 2 (1.7%) cases of cardiac failure were reported for patients who switched from Enbrel to Brenzys treatment. Approximately 15% of subjects who received etanercept achieved an ACR70 response at month 3 and month 6 compared to fewer than 5% of subjects in the placebo arm. Among patients receiving etanercept, the clinical responses generally appeared within 1 to 2 weeks after initiation of therapy and nearly always occurred by 3 months. A dose response was seen; results with 10 mg were intermediate between placebo and 25 mg. Etanercept was significantly better than placebo in all components of the ACR criteria as well as other measures of rheumatoid arthritis disease activity not included in the ACR response criteria, such as morning stiffness. A Health Assessment Questionnaire (HAQ), which included disability, vitality, mental health, general health status and arthritis-associated health status sub-domains, was administered every 3 months during the trial. All sub-domains of the HAQ were improved in patients treated with etanercept compared to controls at 3 and 6 months.
Approximately 15% of subjects who received etanercept achieved an ACR70 response at month 3 and month 6 compared to fewer than 5% of subjects in the placebo arm. Among patients receiving etanercept, the clinical responses generally appeared within 1 to 2 weeks after initiation of therapy and nearly always occurred by 3 months. A dose response was seen; results with 10 mg were intermediate between placebo and 25 mg. Etanercept was significantly better than placebo in all components of the ACR criteria as well as other measures of rheumatoid arthritis disease activity not included in the ACR response criteria, such as morning stiffness. A Health Assessment Questionnaire (HAQ), which included disability, vitality, mental health, general health status and arthritis-associated health status sub-domains, was administered every 3 months during the trial. All sub-domains of the HAQ were improved in patients treated with etanercept compared to controls at 3 and 6 months. In another active-controlled, double-blind, randomised study, clinical efficacy, safety and radiographic progression in RA patients treated with etanercept alone (25 mg twice weekly), methotrexate alone (7.5 to 20 mg weekly, median dose 20 mg) and of the combination of etanercept and methotrexate initiated concurrently were compared in 682 adult patients with active rheumatoid arthritis of 6 months to 20 years duration (median 5 years) who had a less than satisfactory response to at least 1 DMARD other than methotrexate. Forty-three percent of patients had previously received methotrexate a mean of 2 years prior to the trial at a mean dose of 12.9 mg/week. Patients were excluded from this study if methotrexate had been discontinued for lack of efficacy or for safety considerations.
In another active-controlled, double-blind, randomised study, clinical efficacy, safety and radiographic progression in RA patients treated with etanercept alone (25 mg twice weekly), methotrexate alone (7.5 to 20 mg weekly, median dose 20 mg) and of the combination of etanercept and methotrexate initiated concurrently were compared in 682 adult patients with active rheumatoid arthritis of 6 months to 20 years duration (median 5 years) who had a less than satisfactory response to at least 1 DMARD other than methotrexate. Forty-three percent of patients had previously received methotrexate a mean of 2 years prior to the trial at a mean dose of 12.9 mg/week. Patients were excluded from this study if methotrexate had been discontinued for lack of efficacy or for safety considerations. The percentage of patients who achieved low disease activity (defined as DAS < 2.4) at 52 weeks was 39%, 35% and 61% for patients in the etanercept alone group, methotrexate alone group and the etanercept combination group, respectively. Remission (defined as DAS < 1.6) was experienced by 18%, 14% and 37% of patients administered etanercept alone, methotrexate alone and combination therapy, respectively.
The percentage of patients who achieved low disease activity (defined as DAS < 2.4) at 52 weeks was 39%, 35% and 61% for patients in the etanercept alone group, methotrexate alone group and the etanercept combination group, respectively. Remission (defined as DAS < 1.6) was experienced by 18%, 14% and 37% of patients administered etanercept alone, methotrexate alone and combination therapy, respectively. The percentage of patients without progression (TSS change ≤ 0.5) was higher in the etanercept in combination with methotrexate and etanercept groups compared with methotrexate at week 24 (74%, 68% and 56%, respectively; p < 0.05) and week 52 (80%, 68% and 57%, respectively; p < 0.05).
The percentage of patients without progression (TSS change ≤ 0.5) was higher in the etanercept in combination with methotrexate and etanercept groups compared with methotrexate at week 24 (74%, 68% and 56%, respectively; p < 0.05) and week 52 (80%, 68% and 57%, respectively; p < 0.05).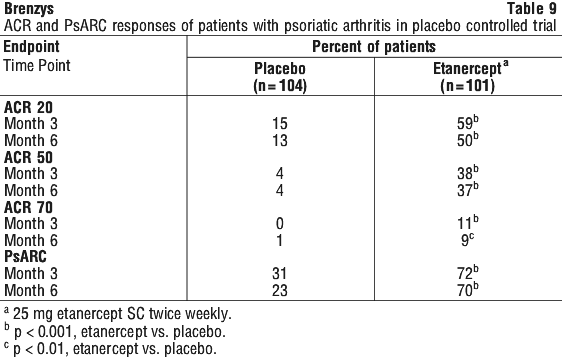 In this study, the psoriatic skin lesions of patients with active arthritis were also improved with etanercept treatment compared with placebo. In a subset of patients with psoriasis involvement ≥ 3% of body surface area, improvements in the Psoriasis Area and Severity Index (PASI) were assessed at Month 3 and Month 6. The PASI is a composite score calculated from disease activity scores and the fraction of body surface area involvement. PASI results are presented in Table 10.
In this study, the psoriatic skin lesions of patients with active arthritis were also improved with etanercept treatment compared with placebo. In a subset of patients with psoriasis involvement ≥ 3% of body surface area, improvements in the Psoriasis Area and Severity Index (PASI) were assessed at Month 3 and Month 6. The PASI is a composite score calculated from disease activity scores and the fraction of body surface area involvement. PASI results are presented in Table 10. Among patients with psoriatic arthritis who received etanercept, the clinical responses were apparent at the time of the first visit (4 weeks) and were maintained through 6 months of therapy. Etanercept was significantly better than placebo in all measures of disease activity (p < 0.001) and responses were similar with and without concomitant methotrexate therapy.
Among patients with psoriatic arthritis who received etanercept, the clinical responses were apparent at the time of the first visit (4 weeks) and were maintained through 6 months of therapy. Etanercept was significantly better than placebo in all measures of disease activity (p < 0.001) and responses were similar with and without concomitant methotrexate therapy. The modified TSS at 6, 12 and 24 months are presented in Table 12 for those patients who entered year 2 and provided radiographs during the second year of the study.
The modified TSS at 6, 12 and 24 months are presented in Table 12 for those patients who entered year 2 and provided radiographs during the second year of the study. In subjects who received placebo during the controlled part of the study and etanercept in the open-label part, further radiographic progression was inhibited after subjects began receiving etanercept. Etanercept treatment resulted in improvement in physical function during the double-blind period and this benefit was maintained during the longer-term exposure of up to 2 years.
In subjects who received placebo during the controlled part of the study and etanercept in the open-label part, further radiographic progression was inhibited after subjects began receiving etanercept. Etanercept treatment resulted in improvement in physical function during the double-blind period and this benefit was maintained during the longer-term exposure of up to 2 years. At 12 weeks, the ASAS 20/50/70 responses were achieved by 60%, 45% and 29%, respectively, of patients receiving etanercept, compared to 27%, 13% and 7%, respectively, of patients receiving placebo (p < 0.001 for etanercept vs placebo). Similar results were seen at week 24. See Table 13.
At 12 weeks, the ASAS 20/50/70 responses were achieved by 60%, 45% and 29%, respectively, of patients receiving etanercept, compared to 27%, 13% and 7%, respectively, of patients receiving placebo (p < 0.001 for etanercept vs placebo). Similar results were seen at week 24. See Table 13.
 At week 12, there was an improvement in the secondary MRI endpoint SPARCC (Spondyloarthritis Research Consortium of Canada) score for the sacroiliac joint for patients receiving etanercept. Adjusted mean change from baseline was -3.8 for etanercept treated (n = 95) versus -0.8 for placebo treated (n = 105) patients.
At week 12, there was an improvement in the secondary MRI endpoint SPARCC (Spondyloarthritis Research Consortium of Canada) score for the sacroiliac joint for patients receiving etanercept. Adjusted mean change from baseline was -3.8 for etanercept treated (n = 95) versus -0.8 for placebo treated (n = 105) patients. The proportions of subjects in the mITT who achieved ASAS 40 were measured at a number of time points in the open label period. At Week 12, 101 subjects who had been randomised to etanercept contributed to the ASAS 40 outcome. By Week 104, 81 subjects who had been randomised to etanercept contributed to the ASAS 40 outcome. Last observation carried forward was used to handle missing values. Based on exploratory analyses, there were no decreases in the proportions of subjects who achieved ASAS 40 at the measurement time points over the open label period compared to Week 12. There are no data on the effects of etanercept on disease progression or structural damage in nr-AxSpA patients. The 2 year data did not reveal any new safety findings.
The proportions of subjects in the mITT who achieved ASAS 40 were measured at a number of time points in the open label period. At Week 12, 101 subjects who had been randomised to etanercept contributed to the ASAS 40 outcome. By Week 104, 81 subjects who had been randomised to etanercept contributed to the ASAS 40 outcome. Last observation carried forward was used to handle missing values. Based on exploratory analyses, there were no decreases in the proportions of subjects who achieved ASAS 40 at the measurement time points over the open label period compared to Week 12. There are no data on the effects of etanercept on disease progression or structural damage in nr-AxSpA patients. The 2 year data did not reveal any new safety findings.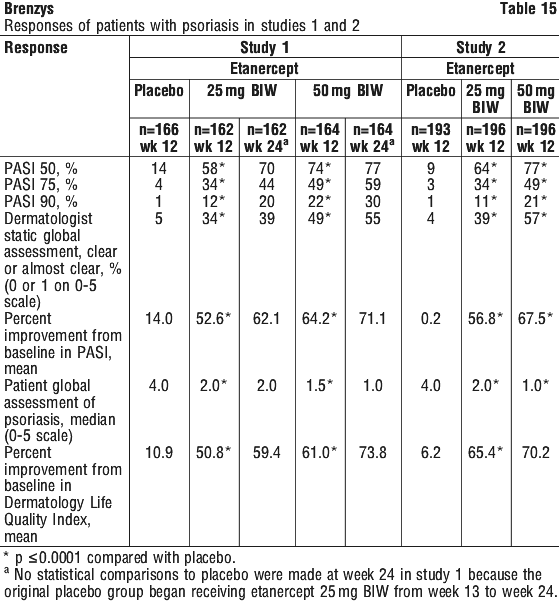 Among patients with plaque psoriasis who received etanercept, significant responses relative to placebo were apparent at the time of the first visit (2 weeks) for the mean percent improvement in PASI, Dermatologist Static Global Assessment of Psoriasis, Dermatology Life Quality Index and Patient Global Assessment of Psoriasis and were maintained through 24 weeks of therapy.
Among patients with plaque psoriasis who received etanercept, significant responses relative to placebo were apparent at the time of the first visit (2 weeks) for the mean percent improvement in PASI, Dermatologist Static Global Assessment of Psoriasis, Dermatology Life Quality Index and Patient Global Assessment of Psoriasis and were maintained through 24 weeks of therapy. Subjects enrolled in either Study 1 or Study 2 (parent studies) were eligible to enter a phase III, open-label study to evaluate the long-term safety, tolerability, and maintenance of efficacy of etanercept in adults with plaque PsO. During the extension study, patients in one arm received etanercept 50 mg once weekly for 48 additional weeks (n = 321). See Figure 6.
Subjects enrolled in either Study 1 or Study 2 (parent studies) were eligible to enter a phase III, open-label study to evaluate the long-term safety, tolerability, and maintenance of efficacy of etanercept in adults with plaque PsO. During the extension study, patients in one arm received etanercept 50 mg once weekly for 48 additional weeks (n = 321). See Figure 6. Etanercept 50 mg once-weekly continued to provide durable efficacy as demonstrated by the percentage of subjects maintaining PASI 50, 75 and 90 responses over time. It was also well tolerated in this population and its safety profile was maintained throughout the extension study.
Etanercept 50 mg once-weekly continued to provide durable efficacy as demonstrated by the percentage of subjects maintaining PASI 50, 75 and 90 responses over time. It was also well tolerated in this population and its safety profile was maintained throughout the extension study. Change in the disease activity score based on a 28 joint count (DAS28) from baseline at Week 24 and Week 52 was also comparable between the treatment groups. The change in DAS28 from baseline at Week 24 was 2.5697 in the Brenzys treatment group and 2.5037 in the EU Enbrel treatment group. The mean change in DAS28 score from baseline at Week 52 was 2.9108 in the Brenzys treatment group and 2.7990 in the EU Enbrel treatment group.
Change in the disease activity score based on a 28 joint count (DAS28) from baseline at Week 24 and Week 52 was also comparable between the treatment groups. The change in DAS28 from baseline at Week 24 was 2.5697 in the Brenzys treatment group and 2.5037 in the EU Enbrel treatment group. The mean change in DAS28 score from baseline at Week 52 was 2.9108 in the Brenzys treatment group and 2.7990 in the EU Enbrel treatment group. Following the 52-week randomised, double blind period (Study SB4-G31-RA), the long-term efficacy, safety/ tolerability, and immunogenicity of Brenzys were assessed in RA patients treated previously with Brenzys or Enbrel. A total of 126 patients from Brenzys treatment group were continued to receive Brenzys (Brenzys/Brenzys) and 119 patients from Enbrel treatment group were switched to Brenzys (Enbrel/Brenzys) in the open-label, extension period (Week 52 to Week 104).
Following the 52-week randomised, double blind period (Study SB4-G31-RA), the long-term efficacy, safety/ tolerability, and immunogenicity of Brenzys were assessed in RA patients treated previously with Brenzys or Enbrel. A total of 126 patients from Brenzys treatment group were continued to receive Brenzys (Brenzys/Brenzys) and 119 patients from Enbrel treatment group were switched to Brenzys (Enbrel/Brenzys) in the open-label, extension period (Week 52 to Week 104).
 Although there is elimination of radioactivity in urine after administration of radiolabelled etanercept to patients and volunteers, increased etanercept concentrations were not observed in patients with acute renal or hepatic failure. The presence of renal and hepatic impairment should not require a change in dosage. There is no apparent pharmacokinetic difference between men and women.
Although there is elimination of radioactivity in urine after administration of radiolabelled etanercept to patients and volunteers, increased etanercept concentrations were not observed in patients with acute renal or hepatic failure. The presence of renal and hepatic impairment should not require a change in dosage. There is no apparent pharmacokinetic difference between men and women.

