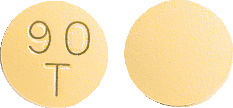
What is in this leaflet
This leaflet answers some of the common questions people ask about BRILINTA. It does not contain all the information that is known about BRILINTA.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor will have weighed the risks of you taking BRILINTA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What BRILINTA is used for
You have been given BRILINTA because you have had:
- a heart attack, or
- unstable angina (angina or chest pain that is not well controlled).
How BRILINTA works
BRILINTA contains a medicine called ticagrelor. This belongs to a group of medicines called anti-platelet medicines.
Platelets are very small cells in your blood that help to stop bleeding. When a blood vessel is damaged, they clump together to help form a blood clot. This stops bleeding.
However, clots can also form inside a damaged blood vessel. This can be very dangerous because:
- the clot can cut off the blood supply completely - this can cause a heart attack (myocardial infarction) or stroke.
- the clot can partly block the blood vessels to the heart - this reduces the blood flow to the heart. This can cause chest pain which comes and goes (called 'unstable angina')
BRILINTA helps stop the clumping of platelets. This reduces the chance of a blood clot forming that can block a blood vessel. This means that BRILINTA reduces the chance of you having another heart attack, chest pain or stroke.
Your doctor will usually also tell you to take acetylsalicylic acid (aspirin). This is another medicine, which affects platelets.
Your doctor will have explained why you are being treated with BRILINTA and told you what dose to take.
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
Your doctor may prescribe this medicine for another use. Ask your doctor if you want more information.
BRILINTA is only available on a doctor's prescription.
There is no evidence that BRILINTA is addictive.
Before you take BRILINTA
When you must not take it
You should not take BRILINTA if:
- you have an allergy to any medicine containing ticagrelor or any of the ingredients listed at the end of the leaflet.
Some of the symptoms of an allergic reaction may include, tightness of the chest, wheezing, coughing or difficulty breathing, swelling of the face, lips, tongue or other parts of the body; rash, itching or hives. - you have problems with bleeding, such as bleeding in your stomach or gut from an ulcer.
- you have moderate to severe liver disease.
- you have had a stroke caused by bleeding in the brain or a history of bleeding in the brain.
- You are taking any of the following medicines:
ketoconazole (used to treat fungal infections), clarithromycin (used to treat bacterial infections), nefazodone (an antidepressant), ritonavir and atazanavir (used to treat HIV infection and AIDS). - the use by (expiry) date printed on the pack has passed or if the packaging is torn or shows signs of tampering.
If you take this product after expiry date has passed, it may not work.
If it has expired or is damaged, return it to your pharmacist for disposal.
Do not take BRILINTA to treat any other complaint unless your doctor advises you to. Do not give this medicine to anyone else.
BRILINTA is not recommended for children under 18 years of age, as its safety and effectiveness in children have not been established.
Do not take BRILINTA if any of the above apply to you. If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor, dentist or pharmacist if:
- you have allergies to any other medicines, foods, preservatives or dyes.
- you have an increased risk of bleeding because of:
- a recent serious injury
- recent surgery (including dental work)
- recent bleeding from your stomach or gut (such as stomach ulcer or colon polyps)
- you have a condition that affects blood clotting - you have asthma or other lung problem or breathing difficulties
- you are due to have surgery (including dental work) at any time while taking BRILINTA.
This is because of the increased risk of bleeding. Your doctor may want you to stop taking BRILINTA for a short time. - you have or have had high uric acid
- you have a low heart rate
- you have or have had any medical conditions.
- you are pregnant or intend to become pregnant.
It is not recommended you take BRILINTA. Your doctor will discuss the risks and benefits of taking BRILINTA during pregnancy. - you are breast feeding or intend to breast feed.
It is not recommended you take BRILINTA. Your doctor will discuss the risks and benefits of taking BRILINTA during breast feeding.
If you have not told your doctor, dentist or pharmacist about any of the above, tell them before you start taking BRILINTA.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Tell your doctor or pharmacist if you are taking any of the following medicines:
- more than 40 mg daily of simvastatin (medicine used to treat high cholesterol)
- rifampin (an antibiotic)
- phenytoin, carbamazepine and phenobarbital (used to control seizures), dexamethasone (used to treat inflammatory and auto immune conditions)
- digoxin (used to treat heart failure)
- cyclosporin (used to lessen your body's defences)
- quinidine and diltiazem (used to treat abnormal heart rhythms)
- adenosine (used to treat irregular heart rate)
- ergotamine (used to treat migraine)
- beta blockers and verapamil (used to treat high blood pressure)
- morphine (used to relieve pain)
Tell your doctor or pharmacist if you are taking any medicines that may increase your risk of bleeding. These medicines include:
- "Oral anticoagulants", often referred to as "blood thinners", which include aspirin, warfarin, clopidogrel and prasugrel.
- "Fibrinolytics" and "thrombolytics", often referred to as "clot-dissolvers", which include streptokinase and tenecteplase.
- Non-steroidal anti-inflammatory drugs (NSAIDs) often taken as pain killers such as ibuprofen and naproxen.
- Selective serotonin reuptake inhibitors (SSRIs) taken as antidepressants such as paroxetine, sertraline and citalopram.
You may need different amounts of your medicines, or may need to take different medicines.
If you are unsure about any medicine you are taking you should check with your doctor or pharmacist. They will have more information on medicines to be careful with or avoid while taking BRILINTA.
How to take BRILINTA
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions in this leaflet, ask your doctor or pharmacist for help.
How to take it
The starting dose is two tablets at the same time. This dose will usually be given to you in the hospital. After that, the usual dose is one tablet twice a day.
BRILINTA film-coated tablets
Swallow each tablet whole with a drink of water.
If you have trouble swallowing BRILINTA film-coated tablets you can crush them and mix with water as follows:
- Crush the tablet(s) to a fine powder using a mortar and pestle or other crushing device.
- Add half a glass of water (approximately 100 mL) to the mortar and pestle/crushing device and stir before pouring the liquid into a glass and drinking immediately.
- For the remaining medicine, add another 100 mL to mortar and pestle/crushing device and stir. Pour the liquid into a glass and drink it immediately. Make sure you also stir the liquid in the glass before you drink it
If you are using a nasogastric tube because you cannot swallow at all:
- Crush tablets in mortar and pestle/crushing device to a fine powder.
- Add 50 mL of water to the mortar and pestle/crushing device and stir before withdrawing the mixture into a syringe and administering the liquid through a nasogastric tube
- For the remaining medicine, add another 50 mL to mortar and pestle/crushing device and stir before withdrawing the mixture into the syringe and administering through a nasogastric tube
- Once all the medicine is used, flush the nasogastric tube with approximately 25 mL of water to deliver any remaining contents of the in the tube into the stomach.
BRILINTA orodispersible tablets
Do not open the blister until it is time to take your medicine.
- To take out the tablet, tear open the blister foil - do not push it through the foil because the tablet may break
- Put the tablet on your tongue and let it disintegrate. Please ensure your hands are dry.
- Once the tablet has disintegrated, you can then swallow it with or without water. Do not use carbonated water.
If you are using a nasogastric tube because you cannot swallow at all:
- Disperse the tablet in water and administer the liquid through the nasogastric tube
- Flush the nasogastric tube with water to deliver any remaining contents of the tube into the stomach.
Your doctor will usually also tell you to take acetylsalicylic acid (aspirin). Your doctor will tell you how much to take (usually 100 mg but may vary between 75-150 mg).
It does not matter whether you take BRILINTA with food or on an empty stomach.
Take your doses at around the same time every day. For example, one in the morning and one in the evening. Taking your tablets at the same time each day will have the best effect. It will also help you to remember when to take it. You can check the day when you last took a BRILINTA film-coated tablet by looking at days on the blister. There is a sun (for the morning) and a moon (for the evening). This will tell you whether you have taken the dose.
How long to take it
You should take BRILINTA for as long as your doctor tells you to.
If you forget to take it
If you forget to take a dose, take your next dose as normal. Then go back to taking it as you would normally.
Do not double the dose (two doses at the same time) to make up for the forgotten dose.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (13 11 26) or go to Accidents and Emergency at your nearest hospital, if you think that you or anyone else may have taken too much BRILINTA even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you take too many BRILINTA tablets you may be at increased risk of bleeding.
While you are taking BRILINTA
Things you must do
- Take BRILINTA exactly as your doctor has told you to.
- Tell all doctors, dentists and pharmacists who are treating you that you are taking BRILINTA.
- If you are about to start taking any new medicines, tell your doctor and pharmacist that you are taking BRILINTA.
- Tell your doctor if you become pregnant while you are taking BRILINTA.
- Tell your doctor if you decide to breast feed your baby.
Things you must not do
- Do not stop taking BRILINTA without talking to your doctor. Take it for as long as your doctor keeps prescribing it. If you want to stop taking BRILINTA, talk to your doctor first.
This is because the benefits of BRILINTA are based on you taking it all the time. If you stop taking BRILINTA without talking to your doctor it may increase the chance of a heart attack or stroke or a blood clot forming. - Do not give this medicine to anyone else, even if they have symptoms that seem similar to yours.
Things to be careful of
Be careful driving or operating machinery. BRILINTA is not likely to affect your ability to drive or use machines. You are being treated because you had a heart attack or unstable angina and you may experience dizziness and confusion. If you have these symptoms, you should be cautious while driving or using machines.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking BRILINTA.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following:
- Feeling short of breath
This is very common (affects more than 1 in 10 people). It might be due to your heart disease or other cause, or it might be a side effect of BRILINTA. If your feeling of shortness of breath gets worse or lasts a long time, tell your doctor. Your doctor will decide if it needs treatment or further investigations. - Bruising
- Nosebleed
- Headache
- Stomach pain
- Constipation, diarrhoea or indigestion
- Feeling of being sick
- Rash
- Inflamed stomach (gastritis)
- A tingling feeling
- Fainting
- Slow and/or irregular heart rate
- Signs of irregular breathing (central sleep apnoea and Cheyne-Stokes respiration)
This has been reported in a small number of patients taking BRILINTA (frequency cannot be estimated from the available data). Central sleep apnoea is associated with irregular breathing and may occur in patients with heart disease, stroke or other causes. Tell your doctor if you develop irregular breathing patterns such as speeding up, slowing down or short pauses in breathing. Your doctor will decide if you need further evaluation.
Tell your doctor immediately if you notice any of the following as you may need urgent medical treatment:
Signs of a stroke, such as:
- sudden numbness or weakness of your arm, leg or face, especially if only on one side of the body.
- sudden confusion, difficulty speaking or understanding others.
- sudden difficulty in walking or loss of balance or co-ordination
- suddenly feeling dizzy or sudden severe headache with no known cause.
These are signs of a kind of stroke caused by bleeding into the brain. This is uncommon (affects less than 1 in 100 people).
Signs of bleeding, including:
- Blood in your urine
- Black stools or blood in your stools
- Bleeding after surgery or from cuts and wounds that is more than normal
Some bleeding is common (affects less than 1 in 10 people). However, severe bleeding is uncommon (affects less than 1 in 100 people), but can be life threatening.
Signs of blood clotting, such as:
- Fever and purplish spots on the skin or in the mouth, with or without yellowing of the skin or eyes, unexplained extreme tiredness or confusion
If any of the following happen, tell your doctor immediately, or go to Accident and Emergency at your nearest hospital:
- tightness of the chest, wheezing, coughing or difficulty breathing.
- swelling of the face, lips, tongue or other parts of the body.
- severe skin reaction which may include rash, itching, redness, blistering or peeling of the skin.
These are very serious side effects. If you have them, you may have had a serious allergic reaction to BRILINTA. You may need urgent medical attention or hospitalisation.
The level of uric acid in your blood may increase while taking BRILINTA. Your doctor may order blood tests to monitor the level of uric acid in your blood.
Tell your doctor if you notice anything else that is making you feel unwell. Some people may get other side effects while taking BRILINTA.
Do not be concerned by this list of side effects. You may not get any of them. If you experience any side effects, do not stop taking BRILINTA until you have spoken with your doctor.
After using it
Storage
Keep your tablets in the blister pack until it is time to take them. If you take BRILINTA out of the blister pack it will not keep well.
Keep it in a cool, dry place where the temperature stays below 30°C.
Do not store it or any other medicine in the bathroom or near a sink. Do not leave it on a windowsill or in the car on hot days. Heat and dampness can destroy some medicines.
Keep it where young children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
Ask your pharmacist what to do with any BRILINTA tablets you have left over if your doctor tells you to stop taking them, or you find that the expiry date has passed.
Product description
What BRILINTA looks like
BRILINTA film-coated tablets
Round, biconvex, yellow, film-coated tablets for oral use with "90" above "T" on one side and plain on the reverse.
Available in blister packs of 14 (not marketed) and 56 tablets.
BRILINTA orodispersible tablets
Round, flat, bevelled edged, white to pale pink, orodispersible tablets for oral use with "90" above "TI" on one side and plain on the reverse.
Available in blister packs of 56 tablets.
Ingredients
BRILINTA film-coated tablets
Each tablet contains ticagrelor 90 mg as the active ingredient.
In addition to ticagrelor, each tablet contains:
- Mannitol
- Calcium hydrogen phosphate dihydrate
- Sodium starch glycollate
- Hydroxypropylcellulose
- Magnesium stearate
- Hyprolose
- Titanium dioxide
- Purified talc
- Macrogol 400
- Iron oxide yellow
BRILINTA orodispersible tablets
Each tablet contains ticagrelor 90 mg as the active ingredient.
In addition to ticagrelor, each tablet contains:
- Mannitol
- Microcrystalline cellulose
- Crospovidone
- Xylitol
- Calcium hydrogen phosphate
- Sodium stearylfumarate
- Hyprolose
- Colloidal anhydrous silica
BRILINTA does not contain gluten.
Supplier
AstraZeneca Pty Ltd
ABN 54 009 682 311
66 Talavera Road
MACQUARIE PARK NSW 2113
Telephone: 1800 805 342
Australian Registration Numbers
BRILINTA 90 mg film-coated tablets - AUST R 167237
BRILINTA 90 mg orodispersible tablets - AUST R 303878
This leaflet was updated in May 2022.
BRILINTA is a registered trade mark of the AstraZeneca group of companies.
© AstraZeneca 2022
Doc ID-002470482 V8.0
Published by MIMS July 2022
 In PLATO, time to first PLATO-defined 'Total Major' bleeding for Brilinta did not differ significantly from that of clopidogrel. The event rate for bleeding was higher for both treatment arms during the first 30 days compared to the remainder of the study; most events occurred during this period. There were few fatal bleeding events in the study, 20 (0.2%) for Brilinta 90 mg twice daily and 23 (0.3%) for clopidogrel 75 mg once daily. When minor bleeding was included, combined PLATO-defined Major and Minor bleeding events were significantly higher on Brilinta than on clopidogrel. Minimal bleeding rates on Brilinta were higher than on clopidogrel. Overall rates of TIMI-defined bleeding events did not differ significantly between Brilinta and clopidogrel.
In PLATO, time to first PLATO-defined 'Total Major' bleeding for Brilinta did not differ significantly from that of clopidogrel. The event rate for bleeding was higher for both treatment arms during the first 30 days compared to the remainder of the study; most events occurred during this period. There were few fatal bleeding events in the study, 20 (0.2%) for Brilinta 90 mg twice daily and 23 (0.3%) for clopidogrel 75 mg once daily. When minor bleeding was included, combined PLATO-defined Major and Minor bleeding events were significantly higher on Brilinta than on clopidogrel. Minimal bleeding rates on Brilinta were higher than on clopidogrel. Overall rates of TIMI-defined bleeding events did not differ significantly between Brilinta and clopidogrel.




 Brilinta is superior to clopidogrel in the prevention of thrombotic events (relative risk reduction [RRR] 16%, absolute risk reduction [ARR] 1.9%, NNT = 54) of the composite efficacy endpoint (CV death, MI, or stroke) over 12 months. The difference in treatments was driven by cardiovascular death and myocardial infarction with no significant difference in the rate of strokes (1.5% on ticagrelor vs 1.3% on clopidogrel). Brilinta demonstrated a statistically significant relative RRR of 16% (ARR 1.1%) for MI and a 21% RRR (ARR 1.1%) for CV death. Treating 91 patients with Brilinta instead of clopidogrel will prevent 1 CV death.
Brilinta is superior to clopidogrel in the prevention of thrombotic events (relative risk reduction [RRR] 16%, absolute risk reduction [ARR] 1.9%, NNT = 54) of the composite efficacy endpoint (CV death, MI, or stroke) over 12 months. The difference in treatments was driven by cardiovascular death and myocardial infarction with no significant difference in the rate of strokes (1.5% on ticagrelor vs 1.3% on clopidogrel). Brilinta demonstrated a statistically significant relative RRR of 16% (ARR 1.1%) for MI and a 21% RRR (ARR 1.1%) for CV death. Treating 91 patients with Brilinta instead of clopidogrel will prevent 1 CV death. Molecular weight: 522.57.
Molecular weight: 522.57.