1 Name of Medicine
Ublituximab.
2 Qualitative and Quantitative Composition
Each vial contains 150 mg of ublituximab in 6 mL at a concentration of 25 mg/mL. The final concentration after dilution is approximately 0.6 mg/mL for the first infusion and 1.8 mg/mL for the second infusion and all subsequent infusions.
Ublituximab is a chimeric monoclonal antibody produced in a clone of the rat myeloma cell line YB2/0 by recombinant DNA technology.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrate for solution for infusion [sterile solution].
Clear to opalescent, and colourless to slightly yellow solution.
The pH of the solution is 6.3 to 6.7, and the osmolality is 340 to 380 mOsm/kg.
4.1 Therapeutic Indications
Briumvi is indicated for the treatment of adult patients with relapsing forms of multiple sclerosis (RMS) with active disease defined by clinical or imaging features.
4.2 Dose and Method of Administration
Treatment should be initiated and supervised by specialised physicians experienced in the diagnosis and treatment of neurological conditions and who have access to appropriate medical support to manage severe reactions such as serious infusion-related reactions (IRRs).
Premedication for infusion-related reactions.
The following two premedications must be administered (orally, intravenously, intramuscular, or subcutaneously) prior to each infusion to reduce the frequency and severity of IRRs (see Section 4.4 Special Warnings and Precautions for Use for additional steps to reduce IRRs):
100 mg methylprednisolone or 10-20 mg dexamethasone (or an equivalent) approximately 30-60 minutes prior to each infusion;
Antihistaminic (e.g. diphenhydramine) approximately 30-60 minutes prior to each infusion.
In addition, premedication with an antipyretic (e.g. paracetamol) may also be considered.
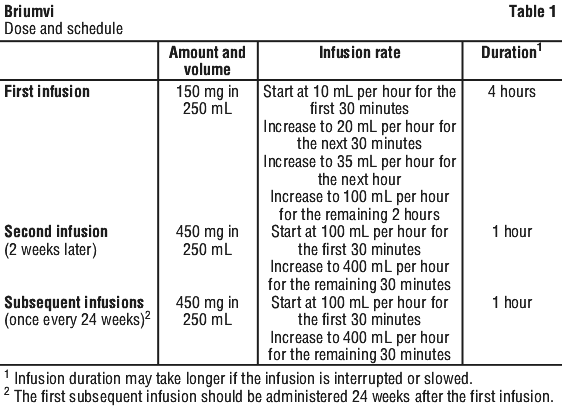
Dose.
First and second doses.
The first dose is administered as a 150 mg intravenous infusion (first infusion), followed by a 450 mg intravenous infusion (second infusion) 2 weeks later (see Table 1).
Subsequent doses.
Subsequent doses are administered as a single 450 mg intravenous infusion every 24 weeks (Table 1). The first subsequent dose of 450 mg should be administered 24 weeks after the first infusion.
A minimal interval of 5 months should be maintained between each dose of ublituximab.
Infusion adjustments in case of IRRs.
Life-threatening IRRs.
If there are signs of a life-threatening or disabling IRR during an infusion, the infusion must be stopped immediately and the patient should receive appropriate treatment. Treatment must be permanently discontinued in these patients (see Section 4.4 Special Warnings and Precautions for Use).
Severe IRRs.
If a patient experiences a severe IRR, the infusion should be interrupted immediately and the patient should receive symptomatic treatment. The infusion should be restarted only after all symptoms have resolved. When restarting, the infusion rate should be at half of the infusion rate at the time of onset of the IRR. If the rate is tolerated, the rate should be increased as described in Table 1.
Mild to moderate IRRs.
If a patient experiences a mild to moderate IRR, the infusion rate should be reduced to half the rate at the onset of the event. This reduced rate should be maintained for at least 30 minutes. If the reduced rate is tolerated, the infusion rate may then be increased as described in Table 1.
Dose modifications during treatment.
No dose reductions are recommended. In case of dose interruption or infusion rate reduction due to IRR, the total duration of the infusion would be increased, but not the total dose.
Delayed or missed doses.
If an infusion is missed, it should be administered as soon as possible; administration after a delayed or missed dose should not wait until the next planned dose. The treatment interval of 24 weeks (with a minimum of 5 months) should be maintained between doses (see Table 1).
Special populations.
Adults over 55 years old and elderly.
Based on the limited data available (see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties), no dose adjustment is considered necessary in patients over 55 years of age.
Renal impairment.
No dose adjustment is expected to be required for patients with renal impairment (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
No dose adjustment is expected to be required for patients with hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Paediatric population.
The safety and efficacy of Briumvi in children and adolescents aged 0 to 18 years have not yet been established. No data are available.
Method of administration.
After dilution, Briumvi is administered as an intravenous infusion through a dedicated line. Infusions should not be administered as an intravenous push or bolus.
 Solutions for intravenous infusion are prepared by dilution of the medicinal product into an infusion bag containing sodium chloride 9 mg/mL (0.9%) solution for injection, to a final concentration of 0.6 mg/mL for the first infusion and 1.8 mg/mL for the second infusion and all subsequent infusions.
Solutions for intravenous infusion are prepared by dilution of the medicinal product into an infusion bag containing sodium chloride 9 mg/mL (0.9%) solution for injection, to a final concentration of 0.6 mg/mL for the first infusion and 1.8 mg/mL for the second infusion and all subsequent infusions.
Instructions for dilution.
Briumvi should be prepared by a healthcare professional using aseptic technique. Do not shake the vial.
The product is intended for single use only.
Do not use the solution if it is discoloured or if it contains foreign particulate matter.
This medicinal product must be diluted before administration. The solution for intravenous administration is prepared by dilution of the product into an infusion bag containing isotonic sodium chloride 9 mg/mL (0.9%) solution for injection.
No incompatibilities between ublituximab and polyvinyl chloride (PVC) or polyolefin (PO) bags and intravenous administration sets have been observed.
For the first infusion, dilute one vial of product into the infusion bag (150 mg/250 mL) to a final concentration of approximately 0.6 mg/mL.
For subsequent infusions, dilute three vials of product into the infusion bag (450 mg/250 mL) to a final concentration of approximately 1.8 mg/mL.
Prior to the start of the intravenous infusion, the content of the infusion bag should be at room temperature (20°C-25°C).
In case an intravenous infusion cannot be completed the same day, the remaining solution should be discarded.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients;
Severe active infection (see Section 4.4 Special Warnings and Precautions for Use);
Patients in a severely immunocompromised state (see Section 4.4 Special Warnings and Precautions for Use);
Known active malignancies.
4.4 Special Warnings and Precautions for Use
Traceability.
In order to improve the traceability of biological medicinal products, the name and the batch number of the administered product should be clearly recorded.
Infusion-related reactions (IRRs).
Symptoms of IRR may include pyrexia, chills, headache, tachycardia, nausea, abdominal pain, throat irritation, erythema, and anaphylactic reaction (see Section 4.8 Adverse Effects (Undesirable Effects)).
Patients should premedicate with a corticosteroid and an antihistamine to reduce the frequency and severity of IRRs (see Section 4.2 Dose and Method of Administration). The addition of an antipyretic (e.g. paracetamol) may also be considered. Patients treated with ublituximab should be observed during infusions. Patients should be monitored for at least one hour after the completion of the first two infusions. Subsequent infusions do not require monitoring post-infusion unless IRR and/or hypersensitivity has been observed. Physicians should inform patients that IRRs can occur up to 24 hours after the infusion.
For guidance regarding posology for patients experiencing IRR symptoms, see Section 4.2 Dose and Method of Administration.
Infection.
Administration must be delayed in patients with an active infection until the infection is resolved.
It is recommended to verify the patient's immune status before dosing since severely immunocompromised patients (e.g. significant neutropenia or lymphopenia) should not be treated (see Section 4.3 Contraindications; Section 4.8 Adverse Effects (Undesirable Effects)).
Ublituximab has the potential for serious, sometimes life-threatening or fatal, infections (see Section 4.8 Adverse Effects (Undesirable Effects)).
Most of the serious infections that occurred in controlled clinical trials in relapsing forms of multiple sclerosis (RMS) resolved. There were 3 infection-related deaths that occurred, all in patients treated with ublituximab; the infections leading to death were post-measles encephalitis, pneumonia, and postoperative salpingitis following an ectopic pregnancy.
Progressive multifocal leukoencephalopathy (PML).
John Cunningham virus (JCV) infection resulting in PML has been observed very rarely in patients treated with anti-CD20 antibodies and mostly associated with risk factors (e.g. patient population, lymphopenia, advanced age, polytherapy with immunosuppressants).
Physicians should be vigilant for the early signs and symptoms of PML, which can include any new onset, or worsening of neurological signs or symptoms, as these can be similar to MS disease.
If PML is suspected, dosing with ublituximab must be withheld. Evaluation including magnetic resonance imaging (MRI) scan preferably with contrast (compared with pre-treatment MRI), confirmatory cerebro-spinal fluid (CSF) testing for JCV deoxyribonucleic acid (DNA) and repeat neurological assessments, should be considered. If PML is confirmed, treatment must be discontinued permanently.
Hepatitis B virus (HBV) reactivation.
HBV reactivation, in some cases resulting in fulminant hepatitis, hepatic failure and death, has been observed in patients treated with anti-CD20 antibodies.
HBV screening should be performed in all patients before initiation of treatment as per local guidelines. Patients with active HBV (i.e. an active infection confirmed by positive results for HBsAg and anti HB testing) should not be treated with ublituximab. Patients with positive serology (i.e. negative for HBsAg and positive for HB core antibody (HBcAb +) or who are carriers of HBV (positive for surface antigen, HBsAg+) should consult liver disease experts before starting the treatment and should be monitored and managed following local medical standards to prevent hepatitis B reactivation.
Vaccinations.
The safety of immunisation with live or live-attenuated vaccines, during or following therapy has not been studied and vaccination with live-attenuated or live vaccines is not recommended during treatment and not until B-cell repletion (see Section 5.1 Pharmacodynamic Properties).
All immunisations should be administered according to immunisation guidelines at least 4 weeks prior to treatment initiation for live or live-attenuated vaccines and, whenever possible, at least 2 weeks prior to treatment initiation for inactivated vaccines.
Vaccination of infants born to mothers treated with ublituximab during pregnancy.
In infants of mothers treated with ublituximab during pregnancy, live or live-attenuated vaccines should not be administered before the recovery of B-cell counts has been confirmed. Depletion of B cells in these infants may increase the risks associated with live or live-attenuated vaccines. Measuring CD19-positive B-cell levels, in neonates and infants, prior to vaccination is recommended.
Inactivated vaccines may be administered as indicated prior to recovery from B-cell depletion. However, assessment of vaccine immune responses, including consultation with a qualified specialist, should be considered to determine whether a protective immune response was mounted.
The safety and timing of vaccination should be discussed with the infant's physician (see Section 4.6 Fertility, Pregnancy and Lactation).
Liver injury.
Clinically significant liver injury, without findings of viral hepatitis, has been reported in the post marketing setting in patients treated with anti-CD20 B-cell depleting therapies approved for the treatment of MS, including Briumvi. Signs of liver injury, including markedly elevated serum hepatic enzymes with elevated total bilirubin, have occurred weeks to months after administration.
Patients treated with Briumvi found to have an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) greater than 3x the upper limit of normal (ULN) with serum total bilirubin greater than 2x ULN, are potentially at risk for severe drug-induced liver injury.
Obtain liver function tests prior to initiating treatment with Briumvi and monitor for signs and symptoms of any hepatic injury during treatment. Measure serum aminotransferases, alkaline phosphatase, and bilirubin levels promptly in patients who report symptoms that may indicate liver injury, including new or worsening fatigue, anorexia, nausea, vomiting, right upper abdominal discomfort, dark urine, or jaundice. If liver injury is present and an alternative etiology is not identified, discontinue Briumvi.
Use in the elderly.
The use of Briumvi in the elderly patient with RMS has not been studied.
Paediatric use.
The safety and efficacy of Briumvi in children and adolescents aged 0 to 18 years have not yet been established. No data are available.
Effects on laboratory tests.
For more information see Section 4.8 Adverse Effects (Undesirable Effects).
Immunoglobulins decrease.
In active-controlled RMS trials, treatment with ublituximab resulted in a decrease in total immunoglobulins over the controlled period of the studies, mainly driven by the reduction in IgM.
Lymphocytes.
In active controlled RMS trials, a transient decrease in lymphocytes was observed in 91% of ublituximab patients at week 1.
Neutrophils counts.
In active-controlled RMS trials, a decrease in neutrophils counts < LLN was observed in 15% of ublituximab patients.4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed.
Vaccinations.
The safety of immunisation with live or live-attenuated vaccines following ublituximab therapy has not been studied, and vaccination with live-attenuated or live vaccines is not recommended during treatment or until B-cell repletion (see Section 4.4 Special Warnings and Precautions for Use; Section 5.1 Pharmacodynamic Properties).
Immunosuppressants.
It is not recommended to use other immunosuppressives concomitantly with ublituximab except corticosteroids for symptomatic treatment of relapses.
When initiating Briumvi after an immunosuppressive therapy, or when initiating an immunosuppressive therapy after Briumvi, the potential for overlapping pharmacodynamic effects should be taken into consideration (see Section 5.1 Pharmacodynamic Properties). Caution should be exercised when prescribing Briumvi taking into consideration the pharmacodynamics of other disease modifying MS therapies.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No studies in animals have been conducted to assess the effects of ublituximab on male or female fertility. No direct adverse effects on male or female reproductive organs were observed at the only dose level evaluated (30 mg/kg/week) in a 26-week intravenous toxicity study in monkeys, which was associated with plasma exposures (AUC) approximately 25 times that in humans at the maximum recommended human dose (450 mg).
(Category C)
Ublituximab is a monoclonal antibody of an immunoglobulin G1 subtype and immunoglobulins are known to cross the placental barrier.
There is a limited amount of data from the use of ublituximab in pregnant women. Postponing vaccination with live or live-attenuated vaccines should be considered for neonates and infants born to mothers who have been exposed to ublituximab during pregnancy. No B-cell count data have been collected in neonates and infants exposed to ublituximab and the potential duration of B-cell depletion in neonates and infants is unknown (see Section 4.4 Special Warnings and Precautions for Use).
Transient peripheral B-cell depletion and lymphocytopenia have been reported in infants born to mothers exposed to other anti-CD20 antibodies during pregnancy.
Briumvi should be avoided during pregnancy unless the potential benefit to the mother outweighs the potential risk to the fetus.
In an enhanced pre- and post-natal development study, pregnant cynomolgus monkeys were administered weekly intravenous doses during organogenesis to parturition of 30 mg/kg ublituximab (corresponding to AUC 24 times the AUC in patients at the maximum recommended dose), which resulted in maternal moribundity and fetal loss. Pathological observations in exposed dams involved multiple organ systems (thrombi in multiple organs, vascular necrosis in the intestine and liver, inflammation and oedema in the lungs and heart) as well as the placenta and these findings were consistent with immune-mediated adverse effects secondary to immunogenicity.
Infant abnormalities were absent in dams exposed during the first trimester of pregnancy. Ublituximab-related external, visceral and skeletal abnormalities were noted in two infants from dams treated during the second trimester of pregnancy. Histopathology evaluations revealed minimal to moderate degeneration/necrosis in the brain. Fetal findings included contractures and abnormal flexion of multiple limbs and tail, shortened mandible, elongate calvarium, enlargement of ears, and/or craniomandibular abnormalities which were attributed to brain necrosis. These findings were potentially related to the immunogenic response of ublituximab in the mothers, which affected the placental exchange of nutrients.
Women of child-bearing potential.
Women of child-bearing potential should use effective contraception while receiving ublituximab and for at least 4 months after the last infusion (see below; see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties).
It is unknown whether ublituximab is excreted in human milk. Human IgGs are known to be excreted in breast milk. Risk to the breast-fed child cannot be excluded.4.7 Effects on Ability to Drive and Use Machines
Briumvi has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
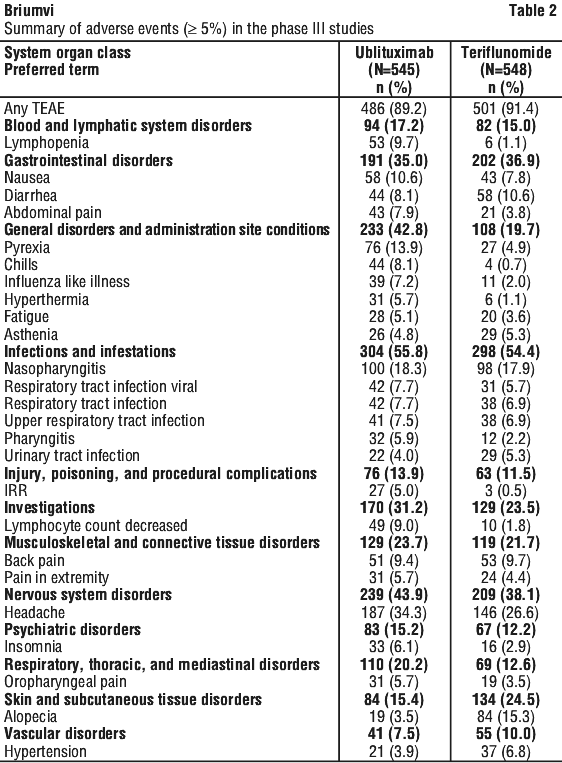
The most important and frequently reported adverse reactions are IRRs (45.3%) and infections (55.8%).
Table 2 summarises the adverse events regardless of causal association that have been reported in ≥ 5% of patients who were treated with either ublituximab or teriflunomide in the phase III studies.
Tabulated list of adverse reactions.
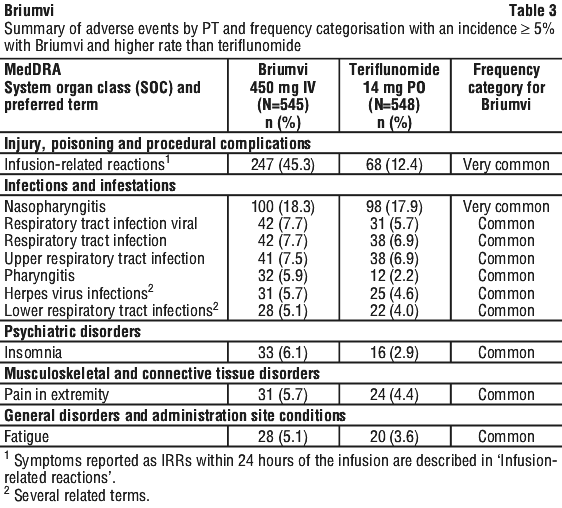
Table 3 summarises the adverse events that have been reported in association with the use of ublituximab. Frequencies are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000) and not known (cannot be estimated from the available data). Within each system organ class and frequency grouping, adverse events are presented in order of decreasing frequency.
Uncommon adverse reactions.
Encephalitis, meningitis, meningoencephalitis.
Frequency not known adverse reactions.
Hepatobiliary disorders: liver injury.
Description of selected adverse reactions.
Infusion-related reactions.
In active-controlled RMS trials, symptoms of IRR included pyrexia, chills, headache, tachycardia, nausea, abdominal pain, throat irritation, erythema, hyperthermia, influenza-like illness, and anaphylactic reaction. IRRs were primarily mild to moderate in severity. The incidence of IRRs in patients treated with ublituximab was 45.3%, with the highest incidence with the first infusion (40.4%). The incidence of IRRs was 8.6% with the second infusion and decreased thereafter. 1.7% of patients experienced IRRs that led to treatment interruption. 0.4% of patients experienced IRRs that were serious. There were no fatal IRRs.
Infection.
In active-controlled RMS trials, the proportion of patients who experienced a serious infection with ublituximab was 5.0% compared to 2.9% in the teriflunomide group. The overall rate of infections in patients treated with ublituximab was similar to patients who were treated with teriflunomide (55.8% vs 54.4%, respectively). The infections were predominantly mild to moderate in severity and consisted primarily of respiratory tract-related infections (mostly nasopharyngitis and bronchitis). Upper respiratory tract infections occurred in 33.6% of ublituximab treated patients and 31.8% teriflunomide treated patients. Lower respiratory tract infections occurred in 5.1% of ublituximab treated patients and 4.0% of teriflunomide treated patients.
Immunoglobulins decrease.
In active-controlled RMS trials, treatment with ublituximab resulted in a decrease in total immunoglobulins over the controlled period of the studies, mainly driven by the reduction in IgM. The proportion of patients at baseline reporting IgG, IgA, and IgM below the lower limit of normal (LLN) in ublituximab treated patients was 6.3%, 0.6%, and 1.1%, respectively. Following treatment, the proportion of ublituximab treated patients reporting IgG, IgA, and IgM below the LLN at 96 weeks was 6.5%, 2.4%, and 20.9%, respectively.
Lymphocytes.
In active controlled RMS trials, a transient decrease in lymphocytes was observed in 91% of ublituximab patients at week 1. The majority of lymphocyte decreases were observed only once for a given patient treated with ublituximab and resolved by week 2 at which time only 7.8% of the patients reported a decrease in lymphocytes. All decreases in lymphocytes were grade 1 (< LLN-800 cells/mm3) and 2 (between 500 and 800 cells/mm3) in severity.
Neutrophils counts.
In active-controlled RMS trials, a decrease in neutrophils counts < LLN was observed in 15% of ublituximab patients compared with 22% of patients treated with teriflunomide. The majority of the neutrophil decreases were transient (only observed once for a given patient treated with ublituximab) and were grade 1 (between < LLN and 1500 cells/mm3) and 2 (between 1000 and 1500 cells/mm3) in severity. Approximately 1% of the patients in the ublituximab group had grade 4 neutropenia vs. 0% in the teriflunomide group. One ublituximab treated patient with grade 4 (< 500 cells/mm3) neutropenia required specific treatment with granulocyte-colony stimulating factor.
Reporting of suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is limited clinical trial experience in RMS with doses higher than the approved intravenous dose of ublituximab. The highest dose tested to date in RMS patients is 600 mg (phase II dose finding study in RMS). The adverse reactions were consistent with the safety profile for ublituximab in the pivotal clinical studies.
There is no specific antidote in the event of an overdose; the infusion should be immediately interrupted and the patient should be observed for IRRs (see Section 4.4 Special Warnings and Precautions for Use).
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: selective immunosuppressants, ATC code: L04AG14.
Mechanism of action.
Ublituximab is a chimeric monoclonal antibody that selectively targets CD20-expressing cells.
CD20 is a cell surface antigen found on pre-B cells, mature and memory B cells but not expressed on lymphoid stem cells and plasma cells. The precise mechanism by which ublituximab exerts its therapeutic effects in multiple sclerosis is unknown, but is presumed to involve binding to CD20 inducing lysis of CD20+ B cells primarily through antibody-dependent cell-mediated cytotoxicity (ADCC) and, to a lesser extent through complement-dependent cytotoxicity (CDC).
Pharmacodynamic effects.
Treatment with ublituximab leads to rapid depletion of CD19+ cells in blood by the first day post treatment as an expected pharmacologic effect. This was sustained throughout the treatment period. For the B cell counts, CD19 is used, as the presence of ublituximab interferes with the recognition of CD20 by the assay.
In the phase III studies, treatment with ublituximab resulted in a median reduction of 97% of CD19+ B cell counts from baseline values after the first infusion in both studies and remained depleted at this level for the duration of dosing.
In the phase III studies, between each dose of ublituximab, 5.5% of patients showed B-cell repletion (> lower limit of normal (LLN) or baseline) at least at one time point.
The longest follow up time after the last ublituximab infusion in the phase III studies indicates that the median time to B-cell repletion (return to baseline/LLN whichever occurred first) was 70 weeks.
Clinical trials.
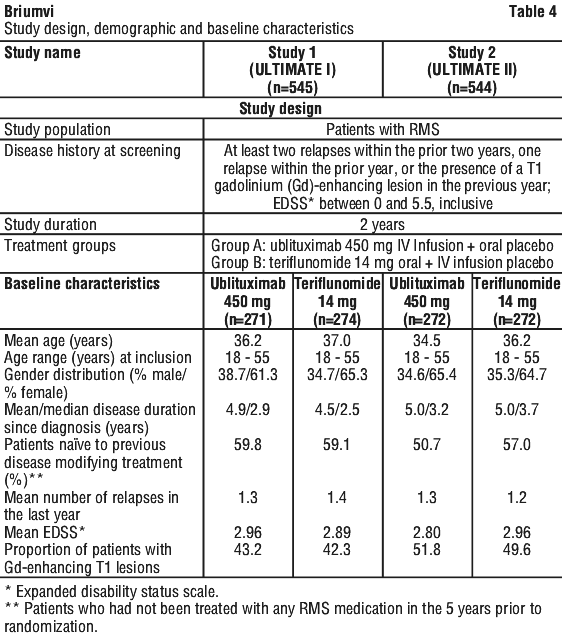
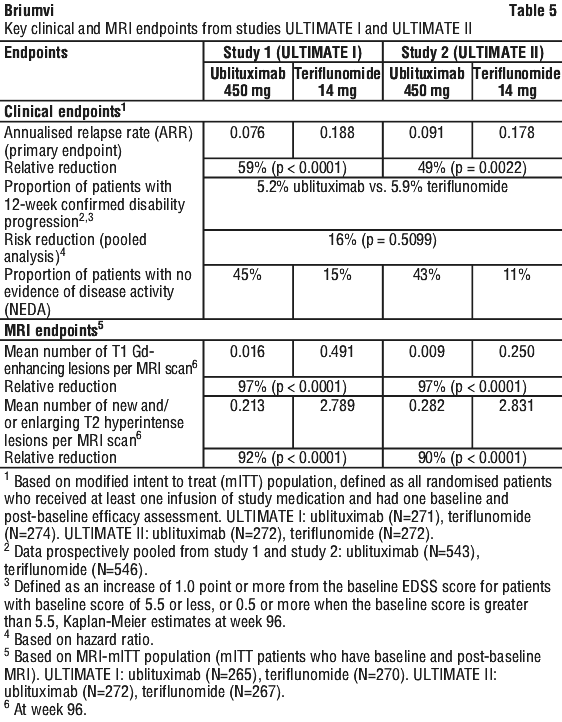
Efficacy and safety of ublituximab were evaluated in two randomised, double-blind, double dummy, active comparator-controlled clinical trials (ULTIMATE I and ULTIMATE II), with identical design, in patients with RMS (in accordance with McDonald criteria 2010) and evidence of disease activity (as defined by clinical or imaging features) within the previous two years. Study design and baseline characteristics of the study population are summarised in Table 4.
Demographic and baseline characteristics were well balanced across the two treatment groups. Patients were to receive either: (1) ublituximab 450 mg plus oral placebo; or (2) teriflunomide 14 mg plus placebo infusion. Oral treatment (active or placebo) was to start on week 1 day 1 and treatment was to continue until the last day of week 95. Infusions (active or placebo) were to begin on week 1 day 1 at 150 mg then increase to 450 mg on week 3 day 15, and continue at 450 mg on week 24, week 48, and week 72.
 Key clinical and MRI efficacy results are presented in Table 5.
Key clinical and MRI efficacy results are presented in Table 5.
The results of these studies show that ublituximab significantly suppressed relapses and subclinical disease activity measured by MRI compared with oral teriflunomide 14 mg.
Immunogenicity.
Serum samples from patients with RMS were tested for antibodies to ublituximab during the treatment period. 81% of ublituximab-treated patients tested positive for anti-drug antibodies (ADA) at one or more timepoints during the 96-week treatment period in clinical efficacy and safety trials. ADA was generally transient (at week 96, 18.5% of patients were positive for ADA). Neutralising activity was detected in 6.4% of ublituximab-treated patients. The presence of ADA or neutralising antibodies had no observable impact on the safety or efficacy of ublituximab.
5.2 Pharmacokinetic Properties
In the RMS studies, the pharmacokinetics (PK) of ublituximab following repeated intravenous infusions was described by a two-compartment model with first-order elimination and with PK parameters typical for an IgG1 monoclonal antibody. Ublituximab exposures increased in a dose-proportional manner (i.e. linear pharmacokinetics) over the dose range of 150 to 450 mg in patients with RMS. Administration of 150 mg ublituximab by intravenous infusion on day 1 followed by 450 mg ublituximab by intravenous infusion over one hour on day 15, week 24 and week 48 led to a geometric mean steady-state AUC of 3000 microgram/mL per day (CV=28%) and a mean maximum concentration of 139 microgram/mL (CV=15%).
Absorption.
Ublituximab is administered as an intravenous infusion. There have been no studies performed with other routes of administration.
Distribution.
In the population pharmacokinetic analysis of ublituximab, the central volume of distribution was estimated to be 3.18 L and the peripheral volume of distribution was estimated to be 3.6 L.
Metabolism.
The metabolism of ublituximab has not been directly studied, as antibodies are cleared principally by catabolism (i.e. breakdown into peptides and amino acids).
Excretion.
Following intravenous infusion of 150 mg ublituximab on day 1 followed by 450 mg ublituximab on day 15, week 24 and week 48, the mean terminal elimination half-life of ublituximab was estimated to be 22 days.
Special populations.
Paediatrics.
No studies have been conducted to investigate the pharmacokinetics of ublituximab in children and adolescents < 18 years of age.
Adults over 55 years old.
There are no dedicated PK studies of ublituximab in patients ≥ 55 years due to limited clinical experience (see Section 4.2 Dose and Method of Administration).
Renal impairment.
No specific studies of ublituximab in patients with renal impairment have been performed. Patients with mild renal impairment were included in the clinical studies. There is no experience in patients with moderate and severe renal impairment. However, as ublituximab is not excreted via urine, it is not expected that patients with renal impairment require dose modification.
Hepatic impairment.
No specific studies of ublituximab in patients with hepatic impairment have been performed.
Since hepatic metabolism of monoclonal antibodies such as ublituximab is negligible, hepatic impairment is not expected to impact its pharmacokinetics. Therefore, it is not expected that patients with hepatic impairment require dose modification.
5.3 Preclinical Safety Data
Genotoxicity.
As a monoclonal antibody, ublituximab is not expected to interact directly with DNA.
Carcinogenicity.
Carcinogenicity studies have not been conducted with ublituximab.6 Pharmaceutical Particulars
6.1 List of Excipients
Sodium chloride; sodium citrate; polysorbate 80; hydrochloric acid (for pH adjustment); water for injections.
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in Section 4.2 Dose and Method of Administration.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
Diluted solution for intravenous infusion.
Chemical and physical in-use stability has been demonstrated for 24 hours at 2°C - 8°C and subsequently for 8 hours at room temperature.
From a microbiological point of view, the prepared infusion should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2°C - 8°C and subsequently for 8 hours at room temperature, unless dilution has taken place in controlled and validated aseptic conditions.
6.4 Special Precautions for Storage
Store at 2°C to 8°C (Refrigerate. Do not freeze.).
Do not shake.
Keep the vial in the outer carton in order to protect from light.
For storage conditions after dilution of the medicinal product, see Section 6.3 Shelf Life.
6.5 Nature and Contents of Container
6 mL concentrate in a glass vial. Pack size of 1 or 3 vials.
6.6 Special Precautions for Disposal
This medicinal product is for single use in one patient only.
In Australia, any unused medicinal product or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.
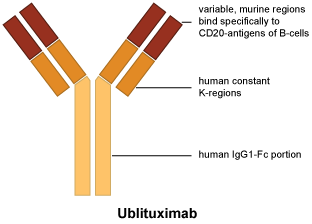
CAS number.
1174014-05-1.7 Medicine Schedule (Poisons Standard)
Schedule 4 (prescription only medicine).
Summary Table of Changes
