1 Name of Medicine
Zanubrutinib.
2 Qualitative and Quantitative Composition
Each hard capsule contains 80 mg zanubrutinib.
Each scored tablet contains 160 mg of zanubrutinib.
Excipients with known effect.
Tablets contain sugars as lactose. For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Capsules.
White to off-white opaque hard capsule of 22 mm in length (size 0), marked with "ZANU 80" in black ink containing white to off-white powder.
Tablets.
Oval, blue, film-coated tablets of 16 mm in length, with letters "zanu" debossed on one side and a functional score line on the other side.
4.1 Therapeutic Indications
Waldenstrom's macroglobulinaemia (WM).
Brukinsa is indicated for the treatment of adult patients with Waldenstrom's macroglobulinaemia (WM) who have received at least one prior therapy, or in first line treatment for patients unsuitable for chemo-immunotherapy.
Mantle cell lymphoma (MCL).
Brukinsa is indicated for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
This indication was approved via the provisional approval pathway, based on objective response rate. Continued approval for this indication depends on verification and description of clinical benefit in the confirmatory trials.
Marginal zone lymphoma (MZL).
Brukinsa is indicated for the treatment of adult patients with marginal zone lymphoma (MZL) who have received at least one-prior anti-CD20-based therapy.
This indication was approved via the provisional approval pathway, based on objective response rate. Continued approval for this indication depends on verification and description of clinical benefit in the confirmatory trials.
Chronic lymphocytic leukaemia (CLL)/small lymphocytic lymphoma (SLL).
Brukinsa is indicated as monotherapy for the treatment of adult patients with chronic lymphocytic leukaemia (CLL) or small lymphocytic lymphoma (SLL), including patients with deletion 17p and/or TP53 mutation.4.2 Dose and Method of Administration
Dosage.
The recommended total daily oral dose of Brukinsa is 320 mg.
Capsules.
Brukinsa capsules may be taken as either 320 mg (four 80 mg capsules) once daily, or as 160 mg (two 80 mg capsules) twice daily.
Tablets.
Brukinsa tablets may be taken as either 320 mg (two 160 mg tablets) once daily or as 160 mg (one 160 mg tablet) twice daily.
Dose modification for adverse reactions.
Recommended dose modifications of Brukinsa for severe (Grade 3) or life-threatening (Grade 4) adverse reactions are provided in Table 1:
 Asymptomatic lymphocytosis should not be regarded as an adverse reaction, and these patients should continue taking Brukinsa.
Asymptomatic lymphocytosis should not be regarded as an adverse reaction, and these patients should continue taking Brukinsa.
Missed dose.
If a dose is not taken at the scheduled time, it can be taken as soon as possible on the same day with a return to the normal schedule the following day.
Method of administration.
Capsules and tablets.
Brukinsa capsules and tablets should be administered orally as 320 mg once daily or 160 mg twice daily approximately every twelve hours. Brukinsa can be taken with or without food. Patients should be instructed to swallow capsules whole, with water. Do not open, break or chew the capsules. Patients should be instructed to swallow tablets whole, with water, but they can be split in half if an 80 mg dose is required. Do not chew or crush the tablets.
Special populations.
Use in children.
The safety and efficacy of Brukinsa have not been established in paediatric patients.
Use in the elderly.
No dosage modification is recommended for elderly patients (aged ≥ 65 years).
Patients with renal insufficiency.
No dosage modification is recommended in patients with mild to moderate renal impairment (creatinine clearance [CrCl] ≥ 30 mL/min, estimated by Cockcroft-Gault). There is limited safety data on patients with severe renal impairment and end-stage renal disease. Monitor for Brukinsa adverse reactions in patients with severe renal impairment (CrCl < 30 mL/min) or on dialysis (see Section 5.2 Pharmacokinetic Properties).
Patients with hepatic insufficiency.
No dosage modification is recommended for patients with mild or moderate hepatic impairment. Based on pharmacokinetic data, the recommended dose of Brukinsa for patients with severe hepatic impairment is 80 mg orally twice daily. The safety of Brukinsa has not been evaluated in patients with severe hepatic impairment. Monitor these patients closely for adverse reactions of Brukinsa (see Section 5.2 Pharmacokinetic Properties).
Interactions requiring dose adjustments.
Dose modification for use with CYP3A inhibitors or inducers (see Table 2):
 After discontinuation of a CYP3A inhibitor or moderate CYP3A inducer, resume previous dose of Brukinsa.
After discontinuation of a CYP3A inhibitor or moderate CYP3A inducer, resume previous dose of Brukinsa.4.3 Contraindications
None.
4.4 Special Warnings and Precautions for Use
Haemorrhage.
Serious and fatal haemorrhagic events have occurred in patients with haematological malignancies treated with zanubrutinib monotherapy. Grade 3 or higher bleeding events occurred in 4.6% of patients, including intracranial and gastrointestinal haemorrhage, haematuria and haemothorax have been reported uncommonly in patients. Bleeding events of any grade occurred in 50.6% of patients with haematological malignancies, including purpura and petechiae.
Bleeding events have occurred in patients taking zanubrutinib with and without concomitant antiplatelet or anticoagulation therapy. Co-administration of Brukinsa with antiplatelet or anticoagulant medications may further increase the risk of haemorrhage and patients should be monitored for signs of bleeding. Discontinue Brukinsa if intracranial haemorrhage of any grade occurs. Consider the benefit-risk of withholding zanubrutinib for 3-7 days pre and post-surgery depending upon the type of surgery and the risk of bleeding.
Infections.
Fatal and non-fatal infections (including bacterial, viral, or fungal infections or sepsis) and opportunistic infections (e.g. herpes viral, cryptococcal, aspergillus, and Pneumocystis jirovecii infections) have occurred in patients with haematological malignancies treated with zanubrutinib monotherapy. Grade 3 or higher infections occurred in these patients. The most common Grade 3 or higher infection was pneumonia. Infections due to hepatitis B virus (HBV) or herpes zoster virus reactivation have occurred.
Consider prophylaxis according to standard of care in patients who are at increased risk for infections. Monitor patients for signs and symptoms of infection and treat appropriately.
Effects on laboratory tests - cytopenias.
Grade 3 or 4 cytopenias including neutropenia, thrombocytopaenia, and anaemia based on laboratory measurements were reported in patients with haematological malignancies treated with zanubrutinib monotherapy (see Section 4.8 Adverse Effects (Undesirable Effects)).
Monitor complete blood counts during treatment. See Section 4.2 Dose and Method of Administration for recommended dose modifications.
Second primary malignancies.
Second primary malignancies, including non-skin carcinoma of any grade have occurred in 14.7% of patients with haematological malignancies treated with zanubrutinib monotherapy. The most frequent second primary malignancy was skin cancer reported in 8.8% of patients (basal cell carcinoma [5.0%], and squamous cell carcinoma of skin [3.5%]). Skin cancers were observed predominantly in patients at high risk of developing skin cancer including those who had medical history of skin cancers. Perform routine skin checks and advise patients to use sun protection. Second primary malignancies of Grade 3 or higher have occurred in 6.6% of patients.
Atrial fibrillation and flutter.
Atrial fibrillation and atrial flutter of any grade have occurred in 4.6% of patients with haematological malignancies treated with zanubrutinib monotherapy, particularly in patients with cardiac risk factors, hypertension, and acute infections. Grade 3 or higher events occurred in 1.9% of patients. Monitor signs and symptoms for atrial fibrillation and atrial flutter and manage as appropriate.
Tumour lysis syndrome.
Tumour lysis syndrome has been infrequently reported with zanubrutinib therapy (< 0.5%), particularly in patients who were treated for chronic lymphocytic leukaemia (CLL). Assess the baseline risk (e.g. high tumour burden) and take appropriate precautions. Monitor patients closely and treat as appropriate.
Paediatric use.
The safety and efficacy of Brukinsa in children below 18 years of age have not been established.
Use in the elderly.
Of the 1550 patients treated with Brukinsa monotherapy, 61.3% were 65 years of age or older. The incidence of Grade 3 or higher adverse events was slightly higher among elderly patients treated with zanubrutinib (69.6% of patients age ≥ 65 versus 62.7% of patients < 65 years of age). No clinically relevant differences in safety were observed between patients ≥ 65 years and younger.4.5 Interactions with Other Medicines and Other Forms of Interactions
Key drug interactions with Brukinsa are summarised in Table 3.

Drug interaction studies.
Agents that may increase zanubrutinib plasma concentrations. CYP3A inhibitors.
The coadministration of multiple doses of itraconazole (strong CYP3A inhibitor) in healthy volunteers increased the Cmax of zanubrutinib by 2.6-fold and AUC by 3.8-fold. The coadministration of multiple doses of strong CYP3A inhibitors voriconazole and clarithromycin in patients with B-cell malignancies resulted in increased zanubrutinib exposures by 3.30-fold and 1.92-fold for dose-normalized AUC0-24 h and 3.29-fold and 2.01-fold for dose-normalized Cmax.
The coadministration of multiple doses of moderate CYP3A inhibitors fluconazole and diltiazem in patients with B-cell malignancies resulted in increased zanubrutinib exposures by 1.88-fold and 1.62-fold for dose-normalized AUC0-24 h and 1.81-fold and 1.62-fold for dose-normalized Cmax.
Concomitant use of zanubrutinib and medicinal products that strongly or moderately inhibit CYP3A can increase zanubrutinib exposure.
Agents that may decrease zanubrutinib plasma concentrations. CYP3A inducers.
Co-administration of multiple doses of rifampicin (strong CYP3A inducer) decreased the zanubrutinib Cmax by 92% and AUC by 93%. Co-administration of multiple doses of rifabutin (moderate CYP3A inducer) decreased the zanubrutinib Cmax by 48% and AUC by 44%.
Concomitant use of zanubrutinib and strong or moderate inducers of CYP3A can decrease zanubrutinib plasma concentrations.
Gastric acid reducing agents.
No clinically significant differences in zanubrutinib pharmacokinetics were observed when co-administered with gastric acid reducing agents (proton pump inhibitors, H2-receptor antagonists).
Agents that may have their plasma concentrations altered by zanubrutinib. CYP3A substrates.
Co-administration of multiple doses of zanubrutinib decreased midazolam (CYP3A substrate) Cmax by 30% and AUC by 47%.
CYP2C19 substrates.
Co-administration of multiple doses of zanubrutinib decreased omeprazole (CYP2C19 substrate) Cmax by 20% and AUC by 36%.
Other CYP substrates.
No clinically significant differences were observed with warfarin (CYP2C9 substrate) pharmacokinetics or predicted with rosiglitazone (CYP2C8 substrate) pharmacokinetics when co-administered with zanubrutinib.
Transporter systems.
Co-administration of multiple doses of zanubrutinib increased digoxin (P-gp substrate) Cmax by 34% and AUC by 11%. No clinically significant differences in the pharmacokinetics of rosuvastatin (BCRP substrate) were observed when co-administered with zanubrutinib.
In vitro studies. CYP enzymes.
Zanubrutinib is a weak inducer of CYP2B6.
Transporter systems.
Zanubrutinib is likely to be a substrate of P-gp. Zanubrutinib is not a substrate or inhibitor of OAT1, OAT3, OCT2, OATP1B1, or OATP1B3.4.6 Fertility, Pregnancy and Lactation
Contraception.
Due to the potential for reproductive toxicity, advise patients who could fall pregnant or who could father a child to avoid doing so by using highly effective contraception (such as condoms) during treatment with zanubrutinib and for at least one week after stopping treatment.
Effects on fertility.
No effect on male or female fertility was noted in rats but at the highest dose tested, morphological abnormalities in sperm and increased post-implantation loss were noted. The high dose of 300 mg/kg/day is approximately 19 and 32 times the human recommended dose in male and female rats, respectively, based on AUC.
(Category D)
There are no clinical data, however, based on findings in animals, zanubrutinib can cause embryofetal harm if administered during pregnancy. Test for pregnancy prior to initiating Brukinsa therapy and inform patients of the potential hazard to a fetus.
Malformations in the heart (2- or 3-chambered hearts) were observed in rats given zanubrutinib at all oral doses of 30, 75 or 150 mg/kg/day during organogenesis, in the absence of maternal toxicity. The lowest dose of 30 mg/kg/day is approximately 4 times the human recommended dose, based on AUC.
Embryotoxicity (post-implantation loss) was observed in rabbits given zanubrutinib during the period of organogenesis at doses of 150 mg/kg/day and was associated with maternal toxicity. The dose of 150 mg/kg/day is approximately 25 times the human recommended dose, based on AUC.
There are no data on the presence of zanubrutinib or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions from zanubrutinib in a breastfed child, advise patients not to breastfeed during treatment with zanubrutinib and for at least two weeks following the last dose.
In a pre- and postnatal developmental toxicity study, zanubrutinib was administered to rats at doses of 30, 75, and 150 mg/kg from implantation through weaning. At doses from 75 mg/kg/day, offspring had decreased body weights pre-weaning. All dose groups had offspring with increased incidences of adverse ocular findings (e.g. cataract, protruding eye). The clinical significance of this is unclear. The dose of 30 mg/kg/day is approximately 4 times the human recommended dose, based on AUC.4.7 Effects on Ability to Drive and Use Machines
No specific studies have been conducted to evaluate the influence of Brukinsa treatment on the ability to drive or operate heavy machinery.
Fatigue, dizziness, and asthenia have been reported in some patients taking Brukinsa and should be considered when assessing a patient's ability to drive or operate machines.
4.8 Adverse Effects (Undesirable Effects)
The safety profile of zanubrutinib monotherapy is based on pooled data from 1550 patients with B-cell malignancies treated with zanubrutinib in 10 clinical trials, including one Phase 1 clinical study (BGB-3111-1002), one Phase 1/2 clinical study (BGB-3111-AU-003), four Phase 2 studies (BGB-3111-205, BGB-3111-206, BGB-3111-210, BGB-3111-214), three Phase 3 clinical studies (BGB-3111-302, BGB-3111-304 and BGB-3111-305) and a long-term extension study (BGB-3111-LTE1). The long-term extension study consists of patients rolling over from studies BGB-3111-AU-003, BGB-3111-205, BGB-3111-206, BGB-3111-210, and BGB-3111-1002. Subjects enrolled in BGB-3111-LTE1 are combined with data from their respective parent studies. Among 1550 patients receiving zanubrutinib, the median duration of exposure was 28.6 months. Among the patients 81% were exposed to zanubrutinib for at least 1 year, 63% were exposed for at least 2 years, and 31% were exposed for at least 3 years.
The most commonly occurring adverse reactions in the 10 studies combined (≥ 20%) were neutropenia+, thrombocytopenia+, upper respiratory tract infection^, bruising^, haemorrhage/haematoma^, musculoskeletal pain, rash^, anaemia+, and pneumonia^. The most common Grade 3 or higher adverse reactions (≥ 5%) were neutropenia+, pneumonia^, thrombocytopenia+, and hypertension.
Discontinuation and dose reduction.
Of the 1550 patients treated with zanubrutinib monotherapy, 59 (3.8%) patients discontinued treatment due to adverse reactions. The most frequent adverse reaction leading to treatment discontinuation was pneumonia§ (1.9%). Adverse reactions leading to dose reduction and dose interruption occurred in 6.1% and 26.9% of patients, respectively.
Table 4 presents adverse reactions that have been reported in association with the use of zanubrutinib monotherapy in the 10 clinical studies. Adverse reactions are listed in Table 4 by MedDRA body system organ class and by frequency. Frequencies are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), not known (cannot be estimated from available data). Within each frequency grouping, adverse reactions are presented in order of decreasing frequency.

Haemorrhage.
Serious and fatal haemorrhagic events have been reported in patients treated with Brukinsa (see Section 4.4 Special Warnings and Precautions for Use).
Infections.
Cases of fatal and non-fatal infections have been reported in patients treated with Brukinsa (see Section 4.4 Special Warnings and Precautions for Use).
Cytopenias.
Cases of neutropenia, anaemia and thrombocytopenia have been reported in patients treated with Brukinsa (see Section 4.4 Special Warnings and Precautions for Use).
Second primary malignancies.
Cases of second primary malignancies have been reported in patients treated with Brukinsa (see Section 4.4 Special Warnings and Precautions for Use).
Atrial fibrillation and flutter.
Cases of atrial fibrillation and flutter have been reported in patients treated with Brukinsa (see Section 4.4 Special Warnings and Precautions for Use).
Tumour lysis syndrome.
Cases of tumour lysis syndrome have been reported in patients with CLL treated with Brukinsa (see Section 4.4 Special Warnings and Precautions for Use).
Waldenstrom's macroglobulinaemia (WM).
The safety of Brukinsa was evaluated in relapsed/refractory (RR) or treatment-naïve WM patients with MYD88 mutation (MYD88MUT) in a Phase 3, randomised, open-label clinical trial, BGB-3111-302, that included 101 patients treated with Brukinsa at a dose of 160 mg twice daily and 98 patients treated with ibrutinib (Cohort 1). Additionally, 28 patients with RR or treatment-naïve WM found to have MYD88 wildtype (MYD88WT) (N=26) or missing/inconclusive MYD88 status (N=2) were treated with Brukinsa in a non-randomised exploratory arm (Cohort 2).
In Cohort 1, the median duration of treatment was 30.3 months in the Brukinsa arm and 29.9 months in the ibrutinib arm. In Cohort 2, the median duration of treatment was 27.8 months.
Serious treatment-emergent adverse events occurred in 48.5% of patients in the Brukinsa arm. The most frequent serious adverse events were febrile neutropenia, influenza, pyrexia, and neutropenia (3% each); and anaemia, pneumonia, basal cell carcinoma, lower respiratory tract infection, pleural effusion, sepsis, and thrombocytopenia (2% each).
Of the 101 patients randomised and treated with Brukinsa, 5% patients discontinued due to adverse events. The events leading to discontinuation were cardiomegaly, neutropenia, plasma cell myeloma, drug-induced liver injury and subdural haemorrhage (1% each). Adverse events leading to dose reduction occurred in 14.9% of patients. The most common adverse events leading to dose reduction were neutropenia (3%) and diarrhoea (2%).
Death due to adverse events within 30 days of last dose occurred in 1 (1%) patient. The adverse event leading to death was cardiomegaly.
Table 5 summarises treatment emergent adverse events in patients randomised in Cohort 1 in BGB-3111-302.
 The safety profile of Brukinsa in patients with WM in the non-randomised Cohort 2 (MYD88WT or missing/inconclusive MYD88 status, N = 28) was generally consistent with the safety profile for Brukinsa in Cohort 1.
The safety profile of Brukinsa in patients with WM in the non-randomised Cohort 2 (MYD88WT or missing/inconclusive MYD88 status, N = 28) was generally consistent with the safety profile for Brukinsa in Cohort 1.
Haematological and chemistry laboratory abnormalities are shown in Table 6.

Mantle cell lymphoma (MCL).
The safety of Brukinsa was evaluated in 118 patients with MCL who received at least one prior therapy at a dose of 320 mg daily in two single-arm clinical trials, BGB-3111-206 and BGB-3111-AU-003. The median duration of treatment was 22.8 months.
Serious treatment-emergent adverse events occurred in 33.9% of patients. The most frequent (≥ 2% of patients) serious adverse events were lung infection (6.8%), pneumonia (4.2%), and anaemia (2.5%).
Of the 118 MCL patients treated with Brukinsa, 13.6% patients discontinued treatment due to adverse events. The most frequent adverse reaction leading to treatment discontinuation was pneumonia (3.4%; grouped terms). Adverse events leading to dose reduction occurred in 3.4% of patients; these included hepatitis B, neutropenia, allergic dermatitis, and peripheral sensory neuropathy (in 1 patient each).
Death due to adverse events within 30 days of last dose occurred in 9 (7.6%) patients. The adverse events leading to death were road traffic accident, cerebral haemorrhage, cerebral infarction, congestive cardiac failure, pneumonia (in 2 patients; grouped terms) and unknown reason (in 3 patients).
Table 7 summarises treatment emergent adverse events in BGB-3111-206 and BGB-3111-AU-003.
 Haematological and chemistry laboratory abnormalities in patients with MCL receiving Brukinsa are summarised in Table 8.
Haematological and chemistry laboratory abnormalities in patients with MCL receiving Brukinsa are summarised in Table 8.

Marginal zone lymphoma (MZL).
The safety of Brukinsa was evaluated in 88 patients with previously treated MZL in two single-arm clinical studies, BGB-3111-214 and BGB-3111-AU-003. Eighty percent received Brukinsa for 6 months or longer, and 67% received treatment for more than one year.
Two fatal adverse events (2.3%) occurred within 30 days of the last dose of Brukinsa, including myocardial infarction and a Covid-19 related death.
Serious adverse events occurred in 40% of patients. The most frequent serious adverse events were pyrexia (8%) and pneumonia (7%).
Adverse events lead to treatment discontinuation in 6% of patients, dose reduction in 2.3%, and dose interruption in 34%. The leading cause of dose modification was respiratory tract infections (13%).
Table 9 summarises treatment emergent adverse events in BGB-3111-214 and BGB-3111-AU-003.
 Table 10 summarises selected laboratory abnormalities.
Table 10 summarises selected laboratory abnormalities.

Chronic lymphocytic leukaemia/small lymphocytic lymphoma (CLL/SLL).
The safety of Brukinsa was evaluated in 938 patients with CLL/SLL across 5 clinical studies: two phase 3 [BGB-3111-304 (SEQUOIA) (n=391) and BGB-3111-305 (ALPINE) (n=324)] and three early phase studies (Phase 2, Phase 1/2 and Phase 1) [Study BGB-3111-205 (n=91), Study BGB-3111-AU-003 (n=123) and Study BGB-3111-1002 (n=9)].
In the Brukinsa group, approximately 66% of patients were ≥ 65 years of age at study entry, which allowed for an adequate assessment of the safety of Brukinsa in older patients. Sixty-six percent of patients in the Brukinsa group were male; 74% of patients were White and 21% were Asian.
Of the 938 patients with CLL/ SLL treated with Brukinsa, adverse events led to treatment discontinuation in 8.0%, dose reduction in 7.5% and dose interruptions in 41.4% of patients. Adverse events leading to death were reported in 36 (3.8%) patients.
SEQUOIA (BGB-3111-304).
The safety of Brukinsa monotherapy was evaluated in previously untreated CLL/SLL patients in a randomised, multicentre, open-label, actively controlled phase 3 trial. Patients without del(17p) mutation (Cohort 1) were randomised to receive either Brukinsa (n=240) until disease progression or unacceptable toxicity or bendamustine plus rituximab (BR) (n = 227) up to 6 cycles. The safety of Brukinsa monotherapy was also evaluated in 111 previously untreated CLL/ SLL patients with del(17p) mutation in a non-randomised single arm (Cohort 2). Patients received Brukinsa monotherapy until disease progression or unacceptable toxicity.
Cohort 1.
The overall median durations of exposure were 6 months and 26 months among patients treated with bendamustine plus rituximab, and Brukinsa, respectively.
Table 11 summarises treatment-emergent adverse events in SEQUOIA (Cohort 1).
 Haematological and chemistry laboratory abnormalities are shown in Table 12.
Haematological and chemistry laboratory abnormalities are shown in Table 12.

Cohort 2.
In this group of previously untreated CLL/SLL patients (n=111) with del(17p) mutation, the overall median treatment duration was 30 months. Serious treatment-emergent adverse events occurred in 45 (40.5%) patients. The most frequent serious treatment-emergent adverse event was pneumonia (6 [5.4%] patients).
The rates of individual treatment-emergent adverse events were similar between Cohorts 1 and 2 i.e. patients without and with del(17p) given Brukinsa, respectively.
ALPINE (BGB-3111-305).
The safety of Brukinsa monotherapy was evaluated in patients with previously treated CLL/SLL in a randomised, multicentre, open-label, Phase 3 actively controlled trial. In ALPINE, 324 patients received Brukinsa monotherapy, and 324 patients received ibrutinib monotherapy until disease progression or unacceptable toxicity.
In ALPINE, the median durations of exposure were 14 months for Brukinsa and 13 months for ibrutinib.
Seventy patients in the Brukinsa arm (22%) reported ≥ 1 serious treatment-emergent adverse event. The most frequent serious adverse event was pneumonia (3.1%).
Table 13 summarises the rates of atrial fibrillation and flutter with Brukinsa vs. ibrutinib based on a pre-specified analysis.
 Table 14 summarises treatment-emergent adverse events in BGB-3111-305 (ALPINE).
Table 14 summarises treatment-emergent adverse events in BGB-3111-305 (ALPINE).
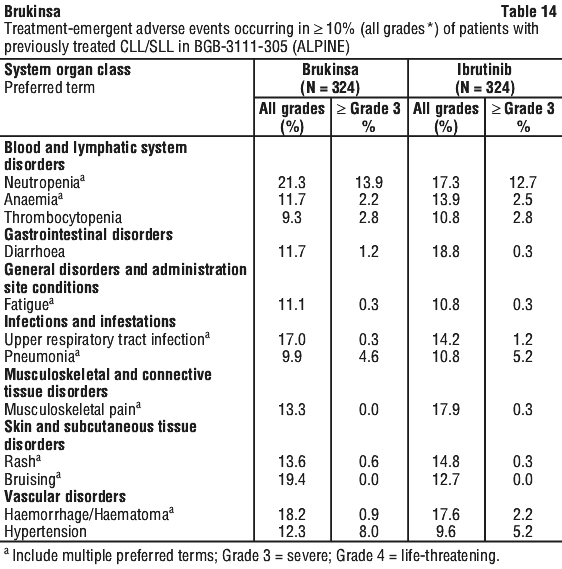 Haematological and chemistry laboratory abnormalities are shown in Table 15.
Haematological and chemistry laboratory abnormalities are shown in Table 15.

Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no specific antidote for Brukinsa. For patients who experience overdose, closely monitor and provide appropriate supportive treatment.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antineoplastic agents, Bruton's tyrosine kinase inhibitors. ATC code: L01EL03.
Mechanism of action.
Zanubrutinib is a small-molecule inhibitor of BTK. Zanubrutinib forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK activity. BTK is a signalling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. In B-cells, BTK signalling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis, and adhesion. In nonclinical studies, zanubrutinib inhibited malignant B-cell proliferation and reduced tumour growth.
Pharmacodynamic effects.
BTK occupancy in peripheral blood mononuclear cells and lymph node biopsies.
The median steady-state BTK occupancy in peripheral blood mononuclear cells was maintained at 100% over 24 hours at a total daily dose of 320 mg in patients with B-cell malignancies. The median steady-state BTK occupancy in lymph nodes was 94% and 100% following the approved recommended dosage of 320 mg once daily, or 160 mg twice daily respectively.
Effect on QT/QTc interval and cardiac electrophysiology.
At the approved recommended doses (320 mg once daily or 160 mg twice daily), there were no clinically relevant effects on the QTc interval. At a single dose 1.5 times the maximum recommended dose (480 mg), zanubrutinib did not prolong the QT interval to any clinically relevant extent (i.e. ≥ 10 msec).
Clinical trials.
Waldenstrom's macroglobulinaemia (WM). BGB-3111-302 (ASPEN): a phase 3, randomized, open-label, multicenter study comparing the efficacy and safety of the Bruton tyrosine kinase inhibitors BGB-3111 and ibrutinib in patients with Waldenstrom's macroglobulinemia.
BGB-3111-302 is a randomised, open-label, multicentre study comparing Brukinsa and ibrutinib in subjects with Waldenstrom's macroglobulinaemia (WM). Eligible patients were at least 18 years of age with a clinical and definite histological diagnosis of relapsed/refractory WM or treatment-naïve when considered by their treating physician to be unsuitable for standard chemo-immunotherapy regimens. Patients had to meet at least one criterion for treatment according to consensus panel criteria from the Seventh International Workshop on Waldenstrom's Macroglobulinaemia (IWWM) and have measurable disease, as defined by a serum IgM level > 0.5 g/dL. Patients with MYD88 mutation (MYD88MUT) were assigned to Cohort 1 (N = 201) and were randomised 1:1 to receive either Brukinsa 160 mg twice daily (Arm A) or ibrutinib 420 mg once daily (Arm B) until disease progression or unacceptable toxicity. Subjects found to have MYD88 wildtype (MYD88WT) by gene sequencing (estimated to be present in approximately 10% of enrolled subjects), were enrolled to Cohort 2 (N = 26) and received Brukinsa 160 mg twice daily on a third, non-randomised, study arm (Arm C). In addition, those subjects whose MYD88 mutational status was missing or inconclusive (N = 2) were assigned to Cohort 2, Arm C.
In Cohort 1 overall, the median age was 70 years (range, 38 to 90 years), 27.9% were > 75 years (22.2% on the ibrutinib arm, 33.3% on the Brukinsa arm), 67% were male, and 91% were Caucasian. At study entry, patients had an International Prognostic Scoring System (IPSS) high categorisation, derived using M-protein by serum protein electrophoresis (SPEP), as follows: 44.4% of patients in the ibrutinib arm and 46.1% of patients in the Brukinsa arm. Ninety-four percent of patients had a baseline ECOG performance status of 0 or 1, and 6.5% had a baseline ECOG performance status of 2. One-hundred-sixty-four patients had relapsed or refractory disease; the median number of prior therapies was 1 (range, 1 to 8). The median time from initial diagnosis was 4.63 years. Overall, 74 (37%) patients had IgM levels ≥ 40 g/L.
In Cohort 2, the median age was 72 years (range, 39 to 87), 42.9% were > 75 years, 50% were male, and 96.4% were Caucasian. At study entry, 42.9% of the patients had an IPSS high categorisation (derived using M-protein by SPEP). Baseline ECOG performance status score was 0 or 1 in 86% of patients and 14% had a baseline ECOG performance status of 2. Twenty-three of the 28 patients in Cohort 2 had relapsed or refractory disease, with a median number of prior therapies of 1 (range, 1 to 5). The median times from initial diagnosis was slightly shorter than in Cohort 1 (median 3.65 years versus 4.6 years). Eight (29%) patients in Cohort 2 had IgM levels ≥ 40 g/L.
In Cohort 1, the primary outcome measure was rate of Complete Response (CR) or Very Good Partial Response (VGPR), as assessed by IRC with adaptation of the response criteria updated at the Sixth IWWM. The secondary endpoints for Cohort 1 include MRR, duration of response, rate of CR or VGPR determined by investigator, PFS, resolution of treatment-precipitating symptoms, and antilymphoma effects in bone marrow and extramedullary disease. The median follow-up was 19.4 months (range 0.5 to 31.1 months) for ibrutinib treated patients and 19.5 months (range 0.4 to 31.2 months) for Brukinsa-treated patients. The study did not meet statistical significance for the pre-specified efficacy outcome of superior CR+VGPR as assessed by IRC, tested first in patients with R/R disease in ASPEN. Results are shown in Table 16.
 In the overall population in Cohort 1, the event-free rates at 12 months for patients in the ibrutinib and Brukinsa treatment arms per overall combined assessment were 87.2% versus 89.7%, respectively, and 83.8% versus 85.0% at 18 months. The event-free rates at 12 months for relapsed/refractory patients in the ibrutinib and Brukinsa treatment arms per overall combined assessment were 85.9% versus 92.4%, respectively, and 81.7% versus 85.9% at 18 months.
In the overall population in Cohort 1, the event-free rates at 12 months for patients in the ibrutinib and Brukinsa treatment arms per overall combined assessment were 87.2% versus 89.7%, respectively, and 83.8% versus 85.0% at 18 months. The event-free rates at 12 months for relapsed/refractory patients in the ibrutinib and Brukinsa treatment arms per overall combined assessment were 85.9% versus 92.4%, respectively, and 81.7% versus 85.9% at 18 months.
Results for Cohort 2 are presented in Table 17.
 In the overall population in Cohort 2, the event-free rates at 12 and 18 months were 72.4% and 68.1%, respectively, per overall combined assessment.
In the overall population in Cohort 2, the event-free rates at 12 and 18 months were 72.4% and 68.1%, respectively, per overall combined assessment.
BGB-3111-AU-003: a phase I/II, open label, multiple dose, dose escalation and expansion study to investigate the safety and pharmacokinetics of the BTK inhibitor BGB 3111 in patients with B-cell lymphoid malignancies.
BGB-3111-AU-003 is a Phase 1/2 open-label, dose-escalation, multicentre, single arm trial of B-cell malignancies including 78 WM patients. Brukinsa was given orally at starting doses ranging from 40 mg daily to 160 mg twice daily until disease progression or unacceptable toxicity. Most patients (93%) received a total daily dose of 320 mg daily (either 320 mg once daily or 160 mg twice daily).
The median age of patients was 67 years (range 40 to 87), 80% were male, and 86% were Caucasian. Ninety-six percent of patients had a baseline ECOG performance status of 0 or 1, and 4% had a baseline ECOG performance status of 2. Fifty-four patients had relapsed or refractory disease; the median number of prior therapies was 2 (range, 1 to 8). The median time from initial diagnosis was 4.31 years. Overall, 24 (31%) patients had IgM levels ≥ 40 g/L.
Seventy-three patients were evaluable for efficacy. Assessment of response was evaluated using the combined response criteria updated at the Sixth IWWM. Results by investigator are shown in Table 18.
 The median durations of VGPR or CR, major response, and overall response have not been reached for the total WM population or relapsed/refractory patients who achieved a response to study treatment.
The median durations of VGPR or CR, major response, and overall response have not been reached for the total WM population or relapsed/refractory patients who achieved a response to study treatment.
The estimated event-free rates at 12, 18, and 24 months for the total WM patient population who achieved a major response were 91.6%, 88.0%, and 83.2%, respectively.
Mantle cell lymphoma (MCL). BGB-3111-206: a single-arm, open-label, multicenter phase 2 study to evaluate efficacy and safety of BGB-3111, a Bruton's tyrosine kinase (BTK) inhibitor, in subjects with relapsed or refractory mantle cell lymphoma (MCL).
BGB-3111-206 is a Phase 2 open-label, multicentre, single arm trial of 86 previously treated MCL patients. Brukinsa was given orally at a dose of 160 mg twice daily until disease progression or unacceptable toxicity.
The median age of patients was 60.5 years (range 34 to 75) and the majority were male (77.9%). The median time since diagnosis was 30 months and the median number of prior therapies was 2 (range 1 to 4). The most common prior regimens were CHOP-based (90.7%) followed by rituximab-based (74.4%). The majority of patients had extranodal involvement (70.9%) and refractory disease (52.3%). Blastoid variant of MCL was present in 14% of patients. The combined biologic MIPI score (which includes age, ECOG score, baseline lactate dehydrogenase, WBC count and Ki-67% staining in tumour cells) was intermediate in 45.3% and high risk in 38.4%.
Tumour response was according to the 2014 Lugano Classification and the primary efficacy endpoint was overall response rate as assessed by an Independent Review Committee (IRC). See Table 19.

BGB-3111-AU-003: a phase I/II, open label, multiple dose, dose escalation and expansion study to investigate the safety and pharmacokinetics of the BTK inhibitor BGB 3111 in patients with B-cell lymphoid malignancies.
BGB-3111-AU-003 is a Phase 1/2 open-label, dose-escalation, multicentre, single arm trial of B-cell malignancies including 37 previously treated MCL patients. Brukinsa was given orally at starting doses ranging from 40 mg daily to 160 mg twice daily until disease progression or unacceptable toxicity. Most patients (32/37) received a total daily dose of 320 mg daily (either 320 mg once daily or 160 mg twice daily).
The median age of patients of the 32 R/R MCL patients receiving 320 mg daily was 70 years (range 42 to 86), and 37.5% of patients were ≥ 75 years old. The majority of patients were male (68.8%). The median time since diagnosis was 4.5 years and the median number of prior therapies was 1 (range 1 to 4). The most common prior regimens were rituximab-based (93.8%) followed by CHOP-based regimen (59.4%). The majority of patients had extranodal involvement (78.1%), and 25% had refractory disease. The MIPI score (which includes age, ECOG score, baseline lactate dehydrogenase and WBC count) was intermediate in 40.6% and high risk in 31.3%.
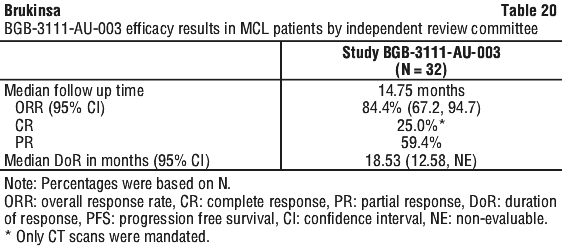
Tumour response was according to the 2014 Lugano Classification and the primary efficacy endpoint was overall response rate as assessed by an Independent Review Committee. PET scans were not required per protocol, and most responses were assessed using CT imaging. See Table 20.
 Marginal zone lymphoma (MZL).
Marginal zone lymphoma (MZL). BGB-3111-214 (MAGNOLIA): a phase 2, open-label study of zanubrutinib (BGB-3111) in patients with relapsed or refractory marginal zone lymphoma; BGB-3111-AU-003: a phase I/II, open label, multiple dose, dose escalation and expansion study to investigate the safety and pharmacokinetics of the BTK inhibitor BGB 3111 in patients with B-cell lymphoid malignancies.
The efficacy of Brukinsa was assessed in Study BGB-3111-214 (MAGNOLIA), a Phase 2 open-label, multicenter, single-arm trial of 68 previously treated patients with MZL who had received at least one prior anti-CD20-based therapy. Twenty-six (38.2%) patients had extranodal MZL, 26 (38.2%) had nodal MZL, 12 (17.6%) had splenic MZL, and 4 (6%) patients had unknown subtype. Brukinsa was given orally at a dose of 160 mg twice daily until disease progression or unacceptable toxicity. The median age of patients was 70 years (range: 37 to 95), and 53% were male. The median time since initial diagnosis was 61.5 months (range: 2.0 to 353.6). The median number of prior treatments was 2 (range: 1 to 6). Twenty-two (32.4%) patients had refractory disease at study entry.
The efficacy of Brukinsa was also assessed in BGB-3111-AU-003, a Phase 1/2, open-label, dose-expansion, global, multicenter, single-arm trial of B-cell malignancies including 20 previously treated MZL patients. Majority of patients (n = 9 [45%]) had extranodal MZL, 6 (30%) had splenic, and 5 (25%) had nodal subtype. Brukinsa was given orally at doses of 160 mg twice daily or 320 mg daily. The median age of patients was 69.5 years (range: 52 to 85). There was equal distribution of male (50%) and female (50%) patients. The median number of prior therapies was 2 (range: 1 to 5).
Tumor response was according to the 2014 Lugano Classification for both studies, and the primary efficacy endpoint was overall response rate as assessed by an IRC. See Table 21.
 In BGB-3111-214 (MAGNOLIA), the median time to response was 2.8 months (range: 1.7 to 11.1 months). The overall response rates were 64%, 76%, 67%, and 50% for the MZL subtypes (extranodal, nodal, splenic, unknown subtype), respectively. The median duration of response was not reached. With further follow-up (median of 23.4 months), the median duration of response was still not reached; however, the lower bound of the 95% CI was estimated at 25.0 months.
In BGB-3111-214 (MAGNOLIA), the median time to response was 2.8 months (range: 1.7 to 11.1 months). The overall response rates were 64%, 76%, 67%, and 50% for the MZL subtypes (extranodal, nodal, splenic, unknown subtype), respectively. The median duration of response was not reached. With further follow-up (median of 23.4 months), the median duration of response was still not reached; however, the lower bound of the 95% CI was estimated at 25.0 months.
In BGB-3111-AU-003, the median time to response was 2.8 months (range: 2.6 to 23.1 months). The overall response rates by MZL subtypes were 89% (extranodal), 100% (nodal), and 50% (splenic).
Chronic lymphocytic leukaemia/small lymphocytic lymphoma (CLL/SLL). The efficacy of Brukinsa in patients with CLL/SLL was evaluated in two randomised controlled trials.
BGB-3111-304 (SEQUOIA): an international, phase 3, open-label, randomized study of BGB 3111 compared with bendamustine plus rituximab in patients with previously untreated chronic lymphocytic leukemia or small lymphocytic lymphoma.
BGB-3111-304 is an ongoing randomised multicentre, open-label, active controlled Phase 3 trial with 4 treatment cohorts in patients with previously untreated CLL/SLL. Cohorts 1 and 2 were included in an interim study report. In Cohort 1 patients without del(17p) were randomised to receive Brukinsa 160 mg bd until disease progression or unacceptable toxicity (Arm A, n=241) or bendamustine (B) 90 mg/m2/day via IV infusion on the first 2 days of each cycle for 6 x 28 day cycles and rituximab (R) 375 mg/m2 via IV infusion for Cycle 1 and 500 mg/m2 for Cycles 2 to 6 (Arm B, n=238). Randomisation in Cohort 1 was stratified by age (< 65 yrs vs. ≥ 65 yrs), Binet stage (C vs. A or B), IGHC mutational status (mutated vs. unmutated) and geographic region (N. America vs. Europe vs. Asia Pacific). In Cohort 2 patients with del(17p) all received open-label Brukinsa (Arm C, n=110) with the same dose regimen as in Arm A.
Demographic and baseline characteristics were generally balanced between Arm A and Arm B of Cohort 1; Arm A had a slightly higher proportion of white patients (91.7%) compared with Arm B (86.6%). In both arms, the median age was 70.0 years, with a slightly higher proportion of patients of ≥ 75 years (26.1%) in Arm A compared with Arm B (22.3%) and a slightly lower proportion of patients 65-75 years old (55.2%) in Arm A compared with Arm B (58.4%).
Demographic and baseline characteristics were generally similar between Arm A in Cohort 1 and Cohort 2 (Arm C). The median age in Cohort 2 was 70.0 years. The proportion of patients 65-75 years old was 55.2% in Arm A and 61.3% in Cohort 2. Arm A included 13.7% and Cohort 2 included 42.3% patients from the Asia Pacific region.
For Cohorts 1 and 2, the primary endpoint was progression-free survival, assessed by an independent central review committee (ICRC) using the 2008 IWCLL guidelines for CLL and the Lugano criteria for SLL. Secondary endpoints included the overall response rate based on ICRC assessment.
In Cohort 1, the median duration of follow-up was 25.0 months (range: 0.0 to 41.4). The estimated progression event-free rate at 24 months was 85.5% (95% CI 80.1, 89.6) for Brukinsa and 69.5% (95% CI 62.4, 75.5) for bendamustine + rituximab (B + R). In Cohort 2, the median duration of follow up was 27.9 (range: 1.0 to 38.8) and the estimated progression event-free rate at 24 months 88.9% (95% CI 81.3, 93.6).
There were 228 (94.6%) patients with an overall response (complete or partial response) in the Brukinsa arm and 203 (85.3%) in the B+R arm in Cohort 1. The estimated 18-month event-free rate in these patients was 91.7% in the Brukinsa arm and 81.3% in the B+R arm. The estimated 24-month event-free rate was 87.5% in the Brukinsa arm and 70.3% in the B+R arm.
Additional efficacy results are presented in Table 22.

BGB-3111-305: a phase 3, randomized study of zanubrutinib (BGB-3111) compared with ibrutinib in patients with relapsed/refractory chronic lymphocytic leukemia or small lymphocytic lymphoma.
BGB-3111-305 is a randomised, multicentre, open-label, Phase 3, active controlled trial. It enrolled 652 patients with relapsed or refractory CLL/SLL after at least one prior systemic therapy. The patients were randomised to either Brukinsa 160 mg orally twice daily or ibrutinib 420 mg orally once daily, continued until disease progression or unacceptable toxicity.
Randomisation was stratified by age (< 65 years versus ≥ 65 years), geographic region (China versus non-China), refractory status (yes or no), and del(17p)/TP53 mutation status (present or absent).
Interim analysis for ORR.
Of 652 patients total, 327 were assigned to Brukinsa monotherapy, 325 to ibrutinib monotherapy. The efficacy evaluation is based on the pre-specified interim analysis of the first 415 randomised patients of the ITT population with median study follow-up of 15.3 months. Of these, 207 were randomised to Brukinsa monotherapy, 208 to ibrutinib monotherapy.
Baseline demographics and disease characteristics were generally balanced between treatment arms in the intent-to-treat (ITT) analysis set and in the first 415 randomised patients. The Brukinsa arm had a higher proportion of female patients compared with the ibrutinib arm (34.9% versus 28.6% in the ITT analysis set and 31.4% versus 25.0% in the first 415 randomised patients). In the ITT analysis set, the median age was 67.0 years in the Brukinsa arm and 68.0 years in the ibrutinib arm, and 67.0 in both arms of the first 415 randomised patients. In both arms of the ITT analysis set 61.5% of patients were ≥ 65 years old. In the first 415 randomized patients, 62.3% of patients in the Brukinsa arm and 61.5% in the ibrutinib arm were ≥ 65 years old. In the ITT analysis set 97.9% of patients in the Brukinsa arm and 96.0% in the ibrutinib arm had an ECOG PS of 0 or 1, and 98.1% and 95.7%, respectively, in the first 415 randomised patients 98.1%.
The primary endpoint was overall response rate (defined as partial response or better) as determined by investigator assessment, using the 2008 IWCLL guidelines for CLL and the Lugano criteria for SLL.
Efficacy results are shown in Table 23.
 Brukinsa demonstrated non-inferiority (1-sided p < 0.0001) and superiority (2-sided p = 0.0006) to ibrutinib in the protocol-specified primary endpoint, overall response rate assessed by investigator.
Brukinsa demonstrated non-inferiority (1-sided p < 0.0001) and superiority (2-sided p = 0.0006) to ibrutinib in the protocol-specified primary endpoint, overall response rate assessed by investigator.
In the total of 652 enrolled patients, the 12-months event-free rates, assessed by independent central review, were 90.4% (95% CI, 85.7, 93.6) for the Brukinsa arm and 81.7% (95% CI, 75.8, 86.4) for the ibrutinib arm.
In patients with del(17p) and/or TP53 mutation in the first 415 randomised patients, the overall response rates were, based on investigator assessment, 80.5% (95% CI 65.1, 91.2; 33 of 41 patients) in the Brukinsa group and 50.0% (95% CI 33.4, 66.6; 19 of 38 patients) in the ibrutinib group. Based on independent review, the overall response rates were 80.5% (95% CI 65.1, 91.2; 33 of 41 patients) in the Brukinsa group and 55.3% (95% CI 38.3, 71.4; 21 of 38 patients) in the ibrutinib group.
Final analysis for ORR.
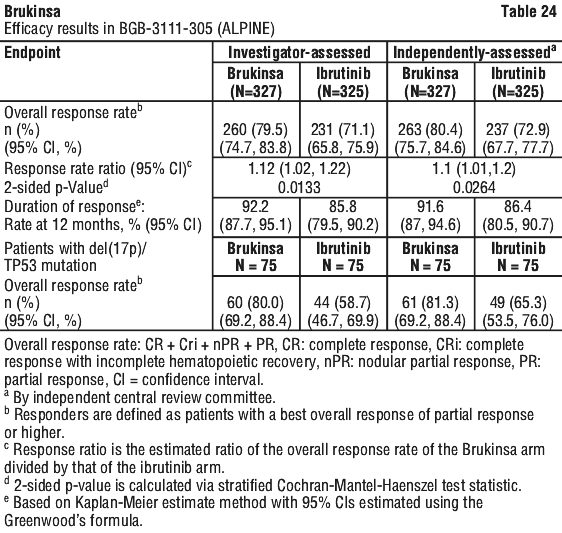
Of the total 652 patients (ITT analysis set), with median follow-up of 24.2 months, Brukinsa demonstrated superiority to ibrutinib in overall response rate by both investigator assessment and independent review. In patients with del(17p) and/or TP53 mutation, overall response rate was also higher in the Brukinsa group. Efficacy results by both investigator and independent review are shown in Table 24.
Final analysis for PFS.
At the prespecified progression-free survival final analysis, with the median study follow-up time of 29.6 months, Brukinsa demonstrated superiority in PFS over ibrutinib (see Table 25).
 See Figure 1.
See Figure 1.
 In patients with del(17p)/TP53 mutation, the hazard ratio for progression-free survival by investigator assessment was 0.53 (95% CI 0.31, 0.88). Based on independent review, the hazard ratio was 0.52 (95% CI 0.30, 0.88). See Figure 2.
In patients with del(17p)/TP53 mutation, the hazard ratio for progression-free survival by investigator assessment was 0.53 (95% CI 0.31, 0.88). Based on independent review, the hazard ratio was 0.52 (95% CI 0.30, 0.88). See Figure 2.
 With an estimated median follow-up of 29.6 months, the median overall survival was not reached in either arm with 17% of patients experiencing an event.
With an estimated median follow-up of 29.6 months, the median overall survival was not reached in either arm with 17% of patients experiencing an event.
5.2 Pharmacokinetic Properties
Zanubrutinib maximum plasma concentration (Cmax) and area under the plasma drug concentration over time curve (AUC) increase proportionally over a dosage range from 40 mg to 320 mg (0.13 to 1 time the recommended total daily dose). Limited systemic accumulation of zanubrutinib was observed following repeated administration. One Brukinsa 160 mg tablet has been demonstrated to be bioequivalent to two Brukinsa 80 mg capsules.
The geometric mean (%CV) zanubrutinib steady-state daily AUC is 2,099 (42%) nanogram.h/mL following a 160 mg twice daily dose and 1,917 (59%) nanogram.h/mL following a 320 mg once daily dose. The geometric mean (%CV) zanubrutinib steady-state Cmax is 299 (56%) nanogram/mL following a 160 mg twice daily dose and 533 (55%) nanogram/mL following a 320 mg once daily dose.
Absorption.
The median Tmax of zanubrutinib is 2 hours. No clinically significant differences in zanubrutinib AUC or Cmax were observed following administration of a high-fat meal (approximately 1,000 calories with 50% of total caloric content from fat) in healthy subjects.
Distribution.
The geometric mean (%CV) apparent steady-state volume of distribution of zanubrutinib during the terminal phase (Vz/F) was 522 (71%) L. The plasma protein binding of zanubrutinib is approximately 94% and the blood-to-plasma ratio was 0.7-0.8.
Metabolism.
Zanubrutinib is primarily metabolised by cytochrome P450(CYP)3A. None of these metabolites are considered to contribute significantly to the safety and efficacy profile of zanubrutinib.
Excretion.
The mean half-life (t½) of zanubrutinib is approximately 2 to 4 hours following a single oral zanubrutinib dose of 160 mg or 320 mg. The geometric mean (%CV) apparent oral clearance (CL/F) of zanubrutinib during the terminal phase was 128 (61%) L/h.
Following a single radio-labelled zanubrutinib dose of 320 mg to healthy subjects, approximately 87% of the dose was recovered in faeces (38% unchanged) and 8% in urine (less than 1% unchanged).
Special populations.
Age (19 to 90 years), sex, ethnicity (Asian, Caucasian, and Other) and body weight (36 to 140 kg) had no clinically meaningful effect on zanubrutinib pharmacokinetics based on population PK analysis.
Renal impairment.
Zanubrutinib undergoes minimal renal elimination. Based on population PK analysis, mild and moderate renal impairment (CrCl ≥ 30 mL/min as estimated by Cockcroft-Gault equation) had no influence on the exposure of zanubrutinib. Limited PK data is available in patients with severe renal impairment (CrCl < 30 mL/min) or in patients requiring dialysis.
Hepatic impairment.
The total AUC of zanubrutinib increased by 11% in subjects with mild hepatic impairment (Child-Pugh class A), by 21% in subjects with moderate hepatic impairment (Child-Pugh class B), and by 60% in subjects with severe hepatic impairment (Child-Pugh class C) relative to subjects with normal liver function. The unbound AUC of zanubrutinib increased by 23% in subjects with mild hepatic impairment (Child-Pugh class A), by 43% in subjects with moderate hepatic impairment (Child-Pugh class B), and by 194% in subjects with severe hepatic impairment (Child-Pugh class C) relative to subjects with normal liver function.
5.3 Preclinical Safety Data
Genotoxicity.
Zanubrutinib was not mutagenic in a bacterial mutagenicity (Ames) assay, was not clastogenic in a chromosome aberration assay in mammalian (CHO) cells, nor was it clastogenic in an in vivo bone marrow micronucleus assay in rats at oral doses up to 2000 mg/kg.
Carcinogenicity.
Carcinogenicity studies have not been conducted with zanubrutinib.6 Pharmaceutical Particulars
6.1 List of Excipients
Capsules.
Capsule content.
Microcrystalline cellulose, croscarmellose sodium, sodium lauryl sulfate, colloidal anhydrous silica, magnesium stearate.
Capsule shell.
Gelatin, titanium dioxide.
Printable ink.
Opacode monogramming ink S-1-277002 Black (ID 107581).
Tablets.
Tablet content.
Lactose monohydrate, croscarmellose sodium, sodium lauryl sulfate, colloidal anhydrous silica, povidone, microcrystalline cellulose, magnesium stearate.
Film coating.
Opadry complete film coating system 00K6505001-CN Blue (ID 140091) containing: hypromellose, titanium dioxide, triacetin, brilliant blue FCF aluminium lake, indigo carmine aluminium lake.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
HDPE bottles with a child-resistant polypropylene closure. Each carton contains one bottle. Each bottle contains either 120 capsules or 60 tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.

CAS number.
1691249-45-2.7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Summary Table of Changes

