





What is in this leaflet
This leaflet answers some common questions about CADIVAST. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking CADIVAST against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What CADIVAST is used for
CADIVAST is used to treat:
- high blood pressure and high cholesterol (fat in the blood).
- angina (a certain type of chest pain).
- people who have high blood pressure and coronary heart disease (CHD) or who are at risk of CHD (for example, if they have diabetes, a history of stroke, or small blood vessel disease). In these people, CADIVAST is used to help reduce the risk of having a heart attack or stroke.
CADIVAST contains a combination of two medicines. One is amlodipine and the other is atorvastatin.
Amlodipine belongs to a group of medicines called calcium channel blockers. They work by relaxing your blood vessels, making it easier for your heart to pump blood around the body and help increase the supply of blood and oxygen to your heart. Calcium channel blockers do not change the amount of calcium in your blood or bones.
Atorvastatin belongs to a group of medicines called statins. It works by reducing the amount of cholesterol made by the liver. It reduces the 'bad' cholesterol and can raise the 'good' cholesterol. Atorvastatin also helps to protect you from a heart attack or stroke.
CADIVAST may be used alone, or in combination with other medicines, to treat your condition.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children.
Before you take CADIVAST
When you must not take it
Do not take CADIVAST if you have an allergy to:
- any medicine containing amlodipine
- any medicine containing atorvastatin
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take CADIVAST if you have active liver disease.
Do not take this medicine if you are pregnant or intend to become pregnant.
If you are a woman of child-bearing age and are taking this medicine, use a proven method of birth control to avoid pregnancy. It may affect your developing baby if you take it during pregnancy.
Do not breast-feed if you are taking this medicine. This medicine may pass into breast milk and there is a possibility that your baby may be affected.
Do not take CADIVAST if you are taking the antibiotic fusidic acid which is used to treat infections
Do not take CADIVAST if you are taking the antivirals glecaprevir/pibrentasvir for the treatment of hepatitis C.
Do not give this medicine to children. Safety and effectiveness in children have not been established.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Your doctor will ask you to have your liver function tested before you start to take CADIVAST.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- heart disease such as heart failure
- liver problems
- kidney problems
- muscle pain, tenderness or weakness from other medicines used to treat cholesterol or triglycerides
- breathing problems
- a type of stroke called a haemorrhagic stroke or a type of stroke called a lacunar stroke.
If you have had one of these strokes before, this medicine may increase the risk of you having a haemorrhagic stroke.
Tell your doctor if you are consuming alcohol regularly.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell him/her before you start taking CADIVAST.
Taking other medicines
Tell your doctor or pharmacist if:
- you are taking other calcium channel blockers. These medicines include amlodipine which is also in CADIVAST; other calcium channel blockers include medicines with the active ingredient felodipine or nifedipine. Check with your doctor or pharmacist if you are unsure.
- you are taking other statins. These medicines include atorvastatin which is also in CADIVAST. Other statins include medicines with active ingredient fluvastatin, pravastatin, rosuvastatin, simvastatin or simvastatin containing medicines. Check with your doctor or pharmacist if you are unsure.
These medicines may be affected by CADIVAST or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
Tell your doctor or pharmacist if you are taking any of the following:
- digoxin, a medicine used to treat some heart problems
- the antibiotics erythromycin, clarithromycin, rifampicin or fusidic acid
- phenytoin, a medicine used to treat epilepsy (seizures)
- oral contraceptives for birth control
- other medicines to treat high cholesterol or triglycerides
- other medicines to treat high blood pressure
- some medicines to treat low potassium
- ciclosporin, tacrolimus, sirolimus or everolimus, medicines used to suppress the immune system
- temsirolimus, a medicine used to treat kidney cancer
- some medicines used to treat some fungal infections, such as ketoconazole or itraconazole
- protease inhibitors for the treatment of HIV infections and/or Hepatitis C, such as efavirenz, fosamprenavir, ritonavir, boceprevir, telaprevir, tipranavir/ritonavir, elbasvir/grazoprevir and simprevir
- HCV non-structural protein 5A (NS5A)/ 5B (NS5B) inhibitors such as daclatasvir and ledipasvir
- letermovir
- diltiazem, a medicine used to treat angina
- antacids, medicines used to treat reflux or ulcers
- spironolactone, a medicine used to treat high blood pressure and certain types of swelling
- vitamin B3
- colchicine, a medicine used to treat a disease with painful swollen joints caused by uric acid crystals
It is also possible that CADIVAST may be affected by some medicines used to treat heart palpitations (or arrhythmias) and St John's Wort.
Your doctor and pharmacist have more information on medicines to be careful with or to avoid while taking CADIVAST.
If you have not told your doctor about any of the above, tell him/her before you start taking CADIVAST.
How to take CADIVAST
Take CADIVAST exactly as your doctor has prescribed.
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
Your doctor may discuss with you the need to be on a diet.
Follow your agreed diet plan carefully.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take
CADIVAST is taken once a day. Your doctor will decide which strength is suitable for you. This will depend on your blood pressure, cholesterol and triglyceride levels.
Your doctor may need to adjust your dose after your blood pressure and the fat levels in your blood have been checked. It is important that you keep your appointments to have these tests done.
How to take it
Swallow the tablets whole with a full glass of water.
Do not crush or chew the tablets.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food.
How long to take it
Take CADIVAST every day and continue taking it for as long as your doctor tells you.
Continue taking your medicine for as long as your doctor tells you.
This medicine to lower cholesterol levels and blood pressure and control the symptoms of angina but it does not cure your condition.
It is important to keep taking your medicine even if you feel well. If you stop taking CADIVAST, your blood pressure and cholesterol levels may rise again.
If you forget to take it
If it is less than 12 hours before your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much CADIVAST. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Keep telephone numbers of these facilities handy.
Symptoms of an overdose may include dizzy, lightheaded or faint and have an irregular heartbeat.
While you are using CADIVAST
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking CADIVAST.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your progress can be checked.
Your cholesterol and triglyceride levels and your liver function tests need to be checked regularly while you are taking this medicine.
A regular blood test to check an enzyme called creatine kinase (CK) may be needed. CK in your blood can rise after muscle injury which can be caused by medicines used to treat cholesterol or triglycerides, such as CADIVAST.
This helps to make sure that CADIVAST is working and to avoid some possible side-effects.
Your blood pressure may also be checked regularly.
Things you must not do
Do not take CADIVAST to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor. If you stop taking it suddenly, your condition may worsen or you may have unwanted side effects such as.
Things to be careful of
Avoid drinking large quantities of alcohol. Drinking large quantities of alcohol while taking CADIVAST may increase your chance of getting liver problems.
Avoid eating large quantities of grapefruit or drinking large quantities of grapefruit juice. Grapefruit juice contains one or more components that alter the metabolism of some medicines, including CADIVAST.
Drinking very large quantities (over 1.2 litres) of grapefruit juice each day while taking CADIVAST increases your chance of getting side effects.
Be careful driving or operating machinery until you know how CADIVAST affects you.
This medicine may cause dizziness or drowsiness in some people and affect alertness.
If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
If you feel light-headed, dizzy or faint when getting out of bed or standing up, get up slowly. Standing up slowly, especially when you get up from bed or chairs, will help your body get used to the change in position and blood pressure. If this problem continues or gets worse, talk to your doctor.
Lifestyle measures that help reduce heart disease risk
By following these simple measures, you can further reduce the risk from heart disease.
- Quit smoking and avoid second-hand smoke.
- Limit alcohol intake.
- Enjoy healthy eating by:
- Eating plenty of vegetables and fruit;
- Reducing your saturated fat intake (eat less fatty meats, full fat dairy products, butter, coconut and palm oils, most take-away foods, commercially-baked products).
- Reducing your salt intake - Be active. Progress, over time, to at least 30 minutes of moderate-intensity physical activity on 5 or more days each week. Can be accumulated in shorter bouts of 10 minutes duration. If you have been prescribed anti-angina medicine, carry it with you when being physically active.
- Maintain a healthy weight.
- Discuss your lifestyle and lifestyle plans with your doctor.
- For more information and tools to improve your heart health, call Heartline, the Heart Foundation's national telephone information service, on 1300 36 27 87 (local call cost).
Know warning signs of heart attack and what to do:
- Tightness, fullness, pressure, squeezing, heaviness or pain in your chest, neck, jaw, throat, shoulders, arms or back.
- You may also have difficulty breathing, or have a cold sweat or feel dizzy or light headed or feel like vomiting (or actually vomit).
- If you have heart attack warning signs that are severe, get worse or last for 10 minutes even if they are mild, call triple zero (000). Every minute counts.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking CADIVAST.
This medicine helps most people with lowering high blood pressure and high cholesterol but it may have unwanted side effects in some people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
If you are over 65 years of age you may have an increased chance of getting side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- headache
- flushing
- dizziness
- tiredness or weakness
- drowsiness or sleepiness
- stomach pain or nausea (feeling sick)
- constipation, diarrhoea, wind
- heartburn, indigestion or wind
- urine infection
- stuffy or runny nose
- nose bleeds
- rash
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor as soon as possible if you notice any of the following:
- swelling of the ankles, feet, face or hands
- muscle and joint pain, muscle weakness, especially in the forearms, thighs, hips, shoulders, neck, and back
- difficulty climbing stairs or standing up from a chair
- difficulty lifting arms over the head
- falling and difficulty getting up from a fall
- symptoms of liver disease such as itching, yellowing of the skin and eyes and dark coloured urine
- feeling weak and tired, excessively thirsty and passing more urine
- dizziness or lightheadedness on standing up from a sitting or lying position
- problems with breathing, including shortness of breath, persistent cough and fever
- eye pain or change in vision
- changes in mood, feeling anxious or nervous
The above list includes serious side effects that may require medical attention.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- symptoms of allergy such as skin rash, itching, swelling of the face, lips, mouth, throat or neck which may cause difficulty in swallowing and breathing
- shortness of breath
- unexpected muscle pain, tenderness or weakness not caused by exercise, particularly if you also feel unwell or have a fever
- tingling in the hands or feet
- changes in heart beat either fast, slow or irregular
- chest pain associated with exertion (angina) that lasts longer, is more severe or occurs more often
- chest pain
- sudden severe headache, which may be accompanied by nausea, vomiting, loss of sensation, tingling in any part of the body or ringing in the ears
- severe blisters and bleeding of the lips, eyes, mouth, nose or genitals
- severe upper stomach pain, often with nausea and vomiting.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
Ask your doctor or pharmacist to answer any questions you may have.
If you are 65 years or older, you should be especially careful while taking CADIVAST. Report any side effects promptly to your doctor.
Some people in this age group maybe more likely to experience side effects such as swelling of the feet and ankles, muscle cramps and dizziness.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some people.
After using CADIVAST
Storage
Keep your tablets in the pack until it is time to take them. If you take the tablets out of the pack they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 25°C.
Do not store CADIVAST or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
CADIVAST tablets are available in eight strengths:
- CADIVAST 5/10 - A white to off-white, film-coated, oval, biconvex tablet debossed with "AA 4" on one side of the tablet and "M" on the other side.
- CADIVAST 5/20 - A white to off-white, film-coated, round, biconvex tablet debossed with "AA 5" on one side of the tablet and "M" on the other side.
- CADIVAST 5/40 - A white to off-white, film-coated, capsule shaped, biconvex tablet debossed with "AA 6" on one side of the tablet and "M" on the other side.
- CADIVAST 5/80 - A white to off-white, film-coated, oval, biconvex tablet debossed with "AA 7" on one side of the tablet and "M" on the other side.
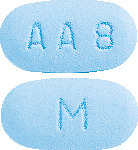
- CADIVAST 10/10 - A blue, film-coated, barrel shaped, biconvex tablet debossed with "AA 8" on one side of the tablet and "M" on the other side.
- CADIVAST 10/20 - A blue, film-coated, oval, biconvex tablet debossed with "AA 9" on one side of the tablet and "M" on the other side.
- CADIVAST 10/40 - A blue, film-coated, round, biconvex tablet debossed with "AA 10" on one side of the tablet and "M" on the other side.
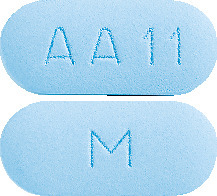
- CADIVAST 10/80 - A blue, film-coated, capsule shaped, biconvex tablet debossed with "AA 11" on one side of the tablet and "M" on the other side.
Each bottle pack contains 30 tablets.
Ingredients
CADIVAST tablets contain the following strength combinations of the active ingredients amlodipine/atorvastatin:
- 5 mg/10 mg
- 5 mg/20 mg
- 5 mg/40 mg
- 5 mg/80 mg
- 10 mg/10 mg
- 10 mg/20 mg
- 10 mg/40 mg
- 10 mg/80 mg
It also contains the following inactive ingredients:
- colloidal anhydrous silica
- sodium carbonate
- microcrystalline cellulose
- arginine
- pregelatinised maize starch
- croscarmellose sodium
- hyprolose
- magnesium stearate
- OPADRY AMB complete film coating system OY-B-28920 White (ARTG PI No: 4271) [5 mg/ 10 mg, 5 mg/20 mg, 5 mg/40 mg and 5 mg/80 mg]
- OPADRY AMB aqueous moisture barrier coating system 80W10932 BLUE (ARTG PI No: 106951) [10 mg/10 mg, 10 mg/20 mg, 10 mg/40 mg and 10 mg/80 mg]
CADIVAST tablets contain soya bean products.
Supplier
CADIVAST is supplied in Australia by:
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was prepared in April 2023.
Australian registration numbers:
CADIVAST 5/10: AUST R 199213
CADIVAST 5/20: AUST R 199222
CADIVAST 5/40: AUST R 199214
CADIVAST 5/80: AUST R 199210
CADIVAST 10/10: AUST R 199215
CADIVAST 10/20: AUST R 199223
CADIVAST 10/40: AUST R 199225
CADIVAST 10/80: AUST R 199217
CADIVAST® is a Viatris company trade mark
CADIVAST_cmi\Apr23/00
Published by MIMS June 2023
 The safety profile of the combination product is consistent with the adverse events previously reported for amlodipine and/or atorvastatin that are detailed below.
The safety profile of the combination product is consistent with the adverse events previously reported for amlodipine and/or atorvastatin that are detailed below. Other adverse experiences which were not clearly dose related but which were reported with an incidence greater than 1.0% in placebo controlled clinical trials include the following (see Table 3).
Other adverse experiences which were not clearly dose related but which were reported with an incidence greater than 1.0% in placebo controlled clinical trials include the following (see Table 3). The following events occurred in ≤ 1% but > 0.1% of patients in controlled clinical trials or under conditions of open trials or marketing experience where a causal relationship is uncertain; they are listed to alert the physician to a possible relationship.
The following events occurred in ≤ 1% but > 0.1% of patients in controlled clinical trials or under conditions of open trials or marketing experience where a causal relationship is uncertain; they are listed to alert the physician to a possible relationship. In the AVALON double blind, placebo controlled study, a total of 847 patients with comorbid hypertension and dyslipidaemia received once daily placebo, 5 mg amlodipine, 10 mg of atorvastatin or the combination of 5 mg amlodipine and 10 mg atorvastatin. The primary objective of the study was the percentage of patients on the combination of amlodipine and atorvastatin reaching JNC VI and NCEP III goals compared to atorvastatin, amlodipine and placebo alone. The results following 8 weeks of treatment are summarised in Table 5. Significantly more patients treated with the combination (45.5%) reached both their blood pressure (BP) and LDL-C goals compared to amlodipine or atorvastatin alone. Amlodipine besilate and atorvastatin calcium trihydrate tablet was not studied in patients with decompensated chronic cardiac failure or postmyocardial infarction (within 3 to 6 months).
In the AVALON double blind, placebo controlled study, a total of 847 patients with comorbid hypertension and dyslipidaemia received once daily placebo, 5 mg amlodipine, 10 mg of atorvastatin or the combination of 5 mg amlodipine and 10 mg atorvastatin. The primary objective of the study was the percentage of patients on the combination of amlodipine and atorvastatin reaching JNC VI and NCEP III goals compared to atorvastatin, amlodipine and placebo alone. The results following 8 weeks of treatment are summarised in Table 5. Significantly more patients treated with the combination (45.5%) reached both their blood pressure (BP) and LDL-C goals compared to amlodipine or atorvastatin alone. Amlodipine besilate and atorvastatin calcium trihydrate tablet was not studied in patients with decompensated chronic cardiac failure or postmyocardial infarction (within 3 to 6 months).
 In a long-term (follow-up at least 6 months, mean 13.8 months) placebo controlled mortality/morbidity study of amlodipine 5 to 10 mg in 1153 patients with NYHA classes III (n = 931) or IV (n = 222) heart failure on stable doses of diuretics, digoxin and angiotensin converting enzyme (ACE) inhibitors, amlodipine had no effect on the primary endpoint of the study which was the combined endpoint of all cause mortality and cardiac morbidity (as defined by life threatening arrhythmia, acute myocardial infarction, or hospitalisation for worsened heart failure), or on NYHA classification, or symptoms of heart failure. Total combined all-cause mortality and cardiac morbidity events were 222/571 (39%) for patients on amlodipine and 246/583 (42%) for patients on placebo: the cardiac morbid events represented about 25% of the endpoints in the study.
In a long-term (follow-up at least 6 months, mean 13.8 months) placebo controlled mortality/morbidity study of amlodipine 5 to 10 mg in 1153 patients with NYHA classes III (n = 931) or IV (n = 222) heart failure on stable doses of diuretics, digoxin and angiotensin converting enzyme (ACE) inhibitors, amlodipine had no effect on the primary endpoint of the study which was the combined endpoint of all cause mortality and cardiac morbidity (as defined by life threatening arrhythmia, acute myocardial infarction, or hospitalisation for worsened heart failure), or on NYHA classification, or symptoms of heart failure. Total combined all-cause mortality and cardiac morbidity events were 222/571 (39%) for patients on amlodipine and 246/583 (42%) for patients on placebo: the cardiac morbid events represented about 25% of the endpoints in the study. In three further trials, 1,148 patients with either heterozygous familial hypercholesterolaemia, non-familial forms of hypercholesterolaemia, or mixed dyslipidaemia were treated with atorvastatin for one year. The results were consistent with those of the dose response study and were maintained for the duration of therapy.
In three further trials, 1,148 patients with either heterozygous familial hypercholesterolaemia, non-familial forms of hypercholesterolaemia, or mixed dyslipidaemia were treated with atorvastatin for one year. The results were consistent with those of the dose response study and were maintained for the duration of therapy. The primary endpoint examined in ASCOT was the rate of fatal coronary heart disease or non-fatal myocardial infarction over 3.3 years. These coronary events occurred in 1.9% of atorvastatin treated patients compared to 3% of placebo treated patients, a relative risk reduction of 36% (p = 0.0005) (Table 8). Although this difference was statistically significant for the whole trial population, this difference was not statistically significant in specified subgroups such as diabetes, patients with left ventricular hypertrophy (LVH), previous vascular disease or metabolic syndrome.
The primary endpoint examined in ASCOT was the rate of fatal coronary heart disease or non-fatal myocardial infarction over 3.3 years. These coronary events occurred in 1.9% of atorvastatin treated patients compared to 3% of placebo treated patients, a relative risk reduction of 36% (p = 0.0005) (Table 8). Although this difference was statistically significant for the whole trial population, this difference was not statistically significant in specified subgroups such as diabetes, patients with left ventricular hypertrophy (LVH), previous vascular disease or metabolic syndrome.

 Chemical name: 3-ethyl, 5-methyl (4RS)-2-[(2-aminoethoxy)methyl]-4-(2-chlorophenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate benzene sulfonate.
Chemical name: 3-ethyl, 5-methyl (4RS)-2-[(2-aminoethoxy)methyl]-4-(2-chlorophenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate benzene sulfonate. Chemical name: (3R, 5R)-7-[2-(4-fluorophenyl)-5-(1-methylethyl)-3-phenyl-4-(phenylcarbamoyl)-1H-pyrrol-1-yl]-3,5-dihydroxyheptanoate calcium trihydrate.
Chemical name: (3R, 5R)-7-[2-(4-fluorophenyl)-5-(1-methylethyl)-3-phenyl-4-(phenylcarbamoyl)-1H-pyrrol-1-yl]-3,5-dihydroxyheptanoate calcium trihydrate.