1 Name of Medicine
Acalabrutinib maleate monohydrate.
2 Qualitative and Quantitative Composition
Each film-coated tablet contains acalabrutinib maleate monohydrate equivalent to 100 mg of acalabrutinib.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
The Calquence 100 mg film-coated tablet is an orange, 7.5 x 13 mm, oval, biconvex tablet, debossed with 'ACA 100' on one side and plain on the reverse.
4.1 Therapeutic Indications
Mantle cell lymphoma (MCL).
Calquence as monotherapy is indicated for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
Calquence in combination with bendamustine and rituximab (BR) is indicated for the treatment of adult patients with previously untreated mantle cell lymphoma (MCL) who are not eligible for autologous haematopoietic stem cell transplantation.
Chronic lymphocytic leukaemia (CLL) / small lymphocytic lymphoma (SLL).
Calquence is indicated for the treatment of patients with chronic lymphocytic leukaemia (CLL)/small lymphocytic lymphoma (SLL).4.2 Dose and Method of Administration
Treatment with Calquence should be initiated and supervised by a physician experienced in the use of anticancer therapies.
Recommended dosage (18 years and above).
Mantle cell lymphoma (MCL).
The recommended dose of Calquence in monotherapy or in combination with other medicines is 100 mg (1 tablet) twice daily. For the combination regimens, refer to the product information of each of the medicine for dosing information. For details of the combination regimens, see Section 5.1 Pharmacodynamic Properties, Clinical trials.
Chronic lymphocytic leukaemia (CLL).
The recommended dose of Calquence for the treatment of CLL is 100 mg (1 tablet) twice daily, either as monotherapy or in combination with obinutuzumab. Administer Calquence prior to obinutuzumab when given on the same day. Refer to the obinutuzumab product information for recommended obinutuzumab dosing information (for details of the combination regimen, see Section 5.1 Pharmacodynamic Properties).
Doses should be separated by approximately 12 hours.
Treatment with Calquence should continue until disease progression or unacceptable toxicity.
Missed dose.
If a patient misses a dose of Calquence by more than 3 hours, they should take the next dose at its regularly scheduled time. Extra tablets of Calquence should not be taken to make up for a missed dose.
Dose adjustments.
Adverse reactions.
Recommended dose modifications for adverse reactions are provided in Table 1 (for patients receiving Calquence monotherapy, or Calquence in combination with obinutuzumab) and Table 2 (for patients receiving Calquence in combination with bendamustine and rituximab). Refer to the product information of each medicine used in combination with Calquence for additional information regarding management of toxicities.


Dose adjustments for use with CYP3A inhibitors or inducers.
Recommended dose adjustments are described in Table 3 (also see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).

Special patient populations.
Renal impairment.
No dose adjustment is recommended in patients with mild to moderate renal impairment (estimated glomerular filtration rate (eGFR) ≥ 30 mL/min/1.73 m2 as estimated by MDRD (modification of diet in renal disease equation)). The pharmacokinetics and safety of Calquence in patients with severe renal impairment (eGFR < 29 mL/min/1.73 m2) or end-stage renal disease have not been studied (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
No dose adjustment is recommended in patients with mild or moderate hepatic impairment (Child-Pugh A, Child-Pugh B, or total bilirubin between 1.5-3 times the upper limit of normal [ULN] and any AST). It is not recommended to administer Calquence in patients with severe hepatic impairment (Child-Pugh C or total bilirubin > 3 times ULN and any AST) (see Section 5.2 Pharmacokinetic Properties).
Severe cardiac disease.
Patients with severe cardiovascular disease were excluded from Calquence clinical studies.
Elderly patients.
No dose adjustment is necessary based on age (see Section 5.2 Pharmacokinetic Properties).
Paediatric patients.
The safety and efficacy of Calquence in children and adolescents aged less than 18 years have not been established.
Method of administration.
Calquence should be swallowed whole with water at approximately the same time each day. Calquence can be taken with or without food. The tablet should not be chewed, crushed, dissolved, or divided.4.3 Contraindications
None.
4.4 Special Warnings and Precautions for Use
Haemorrhage.
Serious haemorrhagic events, including fatal events, have occurred in the combined safety database of 1478 patients with haematological malignancies treated with Calquence monotherapy. Major haemorrhage (Grade 3 or higher bleeding events, serious, or any central nervous system events) occurred in 5.5% of patients, with fatalities occurring in 0.1% of patients. Overall, bleeding events including bruising and petechiae of any grade occurred in 46.1% of patients with haematological malignancies.
The mechanism for the bleeding events is not well understood. Use of antithrombotic agents concomitantly with Calquence may increase the risk of haemorrhage. In Calquence clinical trials, 3% of patients taking Calquence without antithrombotic agents experienced major haemorrhage. The addition of antithrombotic agents increased the percentage to 4.3%. Consider the risks and benefits of antithrombotic agents when co-administered with Calquence. Patients should be monitored for signs of bleeding. Consider the benefit-risk of withholding Calquence for 3-7 days pre- and post-surgery depending upon the type of surgery and the risk of bleeding.
Infection.
Serious infections (bacterial, viral or fungal), including fatal events have occurred in the combined safety database of 1478 patients with haematological malignancies treated with Calquence monotherapy. Consider prophylaxis in patients who are at increased risk for opportunistic infections.
Grade 3 or higher infections occurred in 26.4% of these patients. The most frequently reported Grade 3 or higher infection was pneumonia. Infections due to hepatitis B virus (HBV) reactivation, aspergillosis, and progressive multifocal leukoencephalopathy (PML) have occurred. Monitor patients for signs and symptoms of infection and treat as medically appropriate.
Cytopenias.
In the combined safety database of 1478 patients with haematological malignancies, patients treated with Calquence monotherapy commonly experienced Grade 3 or higher cytopenias, including neutropenia (17.5%), anaemia (9.5%) and thrombocytopenia (6.2%). Monitor complete blood counts as medically appropriate during treatment.
Second primary malignancies.
Second primary malignancies, including non-skin cancers, occurred in 17.6% of patients with haematological malignancies treated with Calquence monotherapy in the combined safety database of 1478 patients. The most frequent second primary malignancy was skin cancer, which occurred in 9.9% of patients. Monitor patients for the appearance of skin cancers. Advise protection from sun exposure.
Atrial fibrillation and flutter.
In the combined safety database of 1478 patients with haematological malignancies treated with Calquence monotherapy, atrial fibrillation and atrial flutter of any grade occurred in 7.4% of patients and Grade 3 or higher occurred in 2.3% of patients. Monitor for symptoms (e.g. palpitations, dizziness, syncope, chest pain, dyspnoea) of atrial fibrillation and atrial flutter and obtain an ECG as appropriate.
Use in the elderly.
Of the 1478 patients in clinical trials of Calquence monotherapy, 42% were ≥ 65 years of age and less than 75 years of age, and 20.6% were 75 years of age or older. No clinically relevant differences in safety or efficacy were observed between patients ≥ 65 years and younger.
Paediatric use.
The safety and efficacy of Calquence in children and adolescents aged less than 18 years have not been established.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Interactions with CYP3A inhibitors and inducers.
The clinical impact and prevention or management of interactions with CYP3A inhibitors or inducers are provided in Table 4. Also see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties.

Effects of acalabrutinib and its active metabolite, ACP-5862, on CYP450 and UGT enzymes.
In vitro data indicate no relevant inhibition of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4/5, UGT1A2 or UGT2B7 by acalabrutinib or ACP-5862 at therapeutic concentrations. Acalabrutinib is a weak inducer of CYP1A2, CYP2B6 and CYP3A4; ACP-5862 weakly induces CYP3A4.
Effects of acalabrutinib and its active metabolite, ACP-5862, on drug transport systems.
Acalabrutinib may increase exposure to co-administered BCRP substrates (e.g. methotrexate) by inhibition of intestinal BCRP.
ACP-5862 may increase exposure to co-administered MATE1 substrates (e.g. metformin) by inhibition of MATE1.
In vitro, acalabrutinib and ACP-5862 are substrates of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Acalabrutinib is not a substrate of renal uptake transporters OAT1, OAT3, and OCT2, or hepatic transporters OATP1B1 and OATP1B3. ACP-5862 is not a substrate of OATP1B1 or OATP1B3. Acalabrutinib and ACP-5862 do not inhibit P-gp, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 and MATE2-K at clinically relevant concentrations.
Effect of food on acalabrutinib.
In healthy subjects, administration of a single 100 mg dose of acalabrutinib tablet with a high fat, high calorie meal (approximately 918 calories, 59 grams carbohydrate, 59 grams fat, and 39 grams protein) did not affect the mean AUC as compared to dosing under fasted conditions. Resulting Cmax decreased by 54% and Tmax was delayed 1-2 hours.
Gastric acid reducing medications.
Acalabrutinib tablets can be co-administered with gastric acid reducing agents (proton pump inhibitors, H2-receptor antagonists, antacids).4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No effects on fertility were observed in male or female rats at exposures 10- or 9-times the clinical exposure (based on AUC) at the recommended dose respectively.
(Category C)
No clinical data are available, but based on findings in animals (see below), Calquence can cause fetal harm and dystocia if administered during pregnancy. Test for pregnancy prior to initiating treatment with Calquence, and advise patients of the potential risk to a fetus. Patients who could become pregnant should use highly effective contraception (such as condoms) during treatment with acalabrutinib and for one week following the last dose.
In an embryofetal study in pregnant rabbits, decreased fetal body weight and delayed skeletal ossification were observed at exposure levels that produced maternal toxicity, which were 2.4 times greater than the clinical exposure (based on AUC) at the recommended dose.
No effects on embryofetal development and survival were observed in one study in pregnant rats, at exposures approximately 9 times greater than the clinical exposure (based on AUC) at the recommended dose. In another study in pregnant rats, underdeveloped renal papilla was observed in F1 generation offspring at doses resulting in maternal exposures approximately 5 times greater than the clinical exposure (based on AUC) at the recommended dose. In two rat reproductive studies, dystocia (prolonged/difficult labour) and mortality of offspring were observed at exposures > 2.3 times greater than the clinical exposure (based on AUC) at the recommended dose. The presence of acalabrutinib and its active metabolite were confirmed in foetal rat plasma. Acalabrutinib and its active metabolite were present in the milk of lactating rats.
No data are available regarding the presence of acalabrutinib or its active metabolite in human milk, its effects on the breastfed child, or on milk production. Acalabrutinib and its active metabolite were present in the milk of lactating rats. Due to the potential for adverse reactions in a breastfed child from Calquence, advise patients not to breastfeed while taking Calquence and for at least 2 weeks after the final dose.4.7 Effects on Ability to Drive and Use Machines
Calquence has no or negligible influence on the ability to drive and use machines. However, during treatment with acalabrutinib fatigue and dizziness have been reported and patients who experience these symptoms should observe caution when driving or using machines.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
Calquence monotherapy.
Across 11 studies, 1478 patients with haematological malignancies have received Calquence as monotherapy at the recommended dose (100 mg twice daily; approximately every 12 hours). The median and mean duration of treatment was 38 months. The most commonly reported (≥ 20%) adverse drug reactions of any grade were infection, diarrhoea, headache, musculoskeletal pain, bruising, cough, arthralgia, fatigue, nausea, leukopenia (mostly neutropenia) and rash. The most commonly reported (≥ 5%) Grade 3 or worse adverse drug reactions were infection, leukopenia, neutropenia, anaemia, second primary malignancy, and thrombocytopenia. Dose reductions due to adverse events were reported in 5.9% of patients (most frequently due to hepatitis B reactivation, sepsis, or diarrhoea). Discontinuation due to adverse events were reported in 14.6% of patients (most frequently due to pneumonia, thrombocytopenia, or diarrhoea).
Calquence in combination regimens.
The safety profile of acalabrutinib when used in combination with other therapies is described specifically for each combination regimen under the relevant clinical trial heading below.
Clinical trials experience.
As clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Safety data from clinical trials in mantle cell lymphoma (MCL).
ECHO (acalabrutinib+BR in previously untreated MCL not eligible for transplant).
The safety of Calquence in combination with bendamustine and rituximab was evaluated in 297 patients with previously untreated MCL in ECHO (see Section 5.1 Pharmacodynamic Properties, Clinical trials). The trial enrolled patients at least 65 years of age with no intention for transplant, with total bilirubin ≤ 1.5 x ULN, AST or ALT ≤ 2.5 x ULN, and estimated creatinine clearance of > 50 mL/min.
The median duration of treatment with Calquence was 28.6 (range: 0.1 to 80) months. A total of 171 (58%) patients were treated with Calquence for ˃ 24 months and 122 (41%) patients were treated for ˃ 36 months.
Serious adverse reactions occurred in 69% of patients who received Calquence. Serious adverse reactions reported in ≥ 2% of patients were COVID-19 (20%), pneumonia (9%), second primary malignancy (7%), pyrexia (5.7%), rash (3.4%), febrile neutropenia (3.4%), atrial fibrillation (3.0%), sepsis (2.7%) and anaemia (2.4%).
Fatal adverse reactions that occurred within 30 days of the last study treatment were reported in 12% of patients who received Calquence plus BR including COVID-19 (6%), pneumonia (1.0%), second primary malignancy (0.7%), sepsis (0.3%), and pneumonitis (0.3%).
Dose interruption, reduction, and discontinuation due to any adverse reaction were reported in 74%, 10% and 43% of patients, respectively. Adverse reactions that resulted in dosage modification in > 10% included infections, cytopenias, rashes, and gastrointestinal toxicity. Adverse reactions which resulted in permanent discontinuation of Calquence in ≥ 4% of patients included COVID-19 and neutropenia.
Table 5 and Table 6 present summaries of the most common adverse reactions and laboratory abnormalities observed in patients with MCL who received Calquence in the ECHO study.
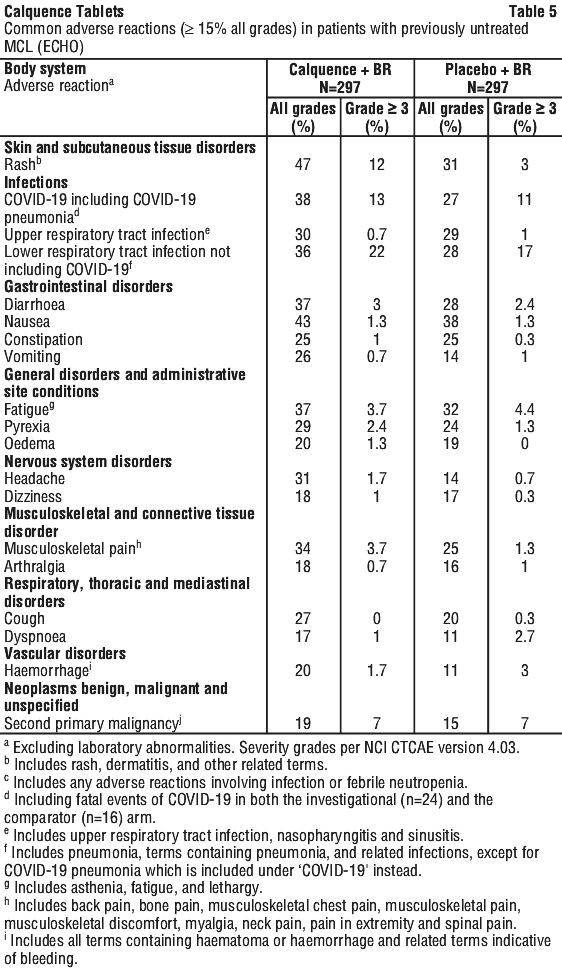 Other clinically relevant adverse reactions (all grades incidence < 15%) in patients receiving Calquence + BR included: bruising (14%), abdominal pain (12%), atrial fibrillation or flutter (7%), and tumour lysis syndrome (1.3%).
Other clinically relevant adverse reactions (all grades incidence < 15%) in patients receiving Calquence + BR included: bruising (14%), abdominal pain (12%), atrial fibrillation or flutter (7%), and tumour lysis syndrome (1.3%).

ACE-LY-004 (acalabrutinib monotherapy in previously treated MCL).
The safety data described in this section reflect exposure to Calquence (100 mg twice daily) in 124 patients with previously treated MCL in ACE-LY-004 (see Section 5.1 Pharmacodynamic Properties, Clinical trials). The median duration of treatment with Calquence was 16.6 (range 0.1 to 26.6) months. A total of 91 (73%) patients were treated with Calquence for ≥ 6 months and 74 (60%) patients were treated for ≥ 1 year.
The most common adverse reactions (≥ 20%) of any grade were anaemia, thrombocytopenia, headache, neutropenia, diarrhoea, fatigue, myalgia, and bruising. Grade 1 severity for the non-haematologic, most common events were as follows: headache (25%), diarrhoea (16%), fatigue (20%), myalgia (15%), and bruising (19%). The most common Grade ≥ 3 non-haematological adverse reaction (reported in at least 2% of patients) was diarrhoea.
Dose reductions and discontinuation due to any adverse reaction were reported in 1.6% and 6.5% of patients, respectively.
Table 7 and Table 8 present the frequency category of adverse reactions observed in patients with MCL treated with Calquence.

 Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 4.8% of patients.
Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 4.8% of patients.
Safety data from clinical trials in chronic lymphocytic leukaemia (CLL).
The safety data described below reflect exposure to Calquence (100 mg twice daily) in two randomised controlled clinical trials (ELEVATE-TN and ASCEND) in patients with CLL (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
The most common adverse reactions (≥ 20%) of any grade were infection, neutropenia, anaemia, thrombocytopenia, headache, diarrhoea, musculoskeletal pain, bruising, and nausea. The most commonly reported Grade ≥ 3 adverse reactions were infection, neutropenia, and anaemia.
ELEVATE-TN (acalabrutinib+obinutuzumab+chlorambucil in previously untreated CLL). The safety of Calquence plus obinutuzumab (Calquence+G), Calquence monotherapy, and obinutuzumab plus chlorambucil (GClb) was evaluated in a randomised, multicentre, open-label, Phase 3 study, in 526 patients with previously untreated CLL. Details of the study treatment are described in Section 5.1 Pharmacodynamic Properties, Clinical trials.
In the Calquence+G arm, adverse events led to regimen discontinuation in 11% of patients and a dose reduction of Calquence in 8% of patients. In the Calquence monotherapy arm, adverse events led to discontinuation in 9% and dose reduction in 3% of patients. In the GClb arm, adverse events led to regimen discontinuation in 14% of patients and a dose reduction of chlorambucil in 28% of patients. There were no dose reductions for obinutuzumab.
The adverse reactions described in Table 9 and Table 10 reflect exposure to Calquence in the Calquence+G and Calquence monotherapy arms with a median duration of exposure of 27.7 months in patients with previously untreated CLL. The median duration of exposure in the GClb arm was 5.6 months.


Tumour lysis syndrome in ELEVATE-TN.
Tumour lysis syndrome (TLS) was reported in 2% of patients treated with Calquence+G. No patients experienced TLS in the Calquence monotherapy arm.
Atrial fibrillation/atrial flutter in ELEVATE-TN.
Atrial fibrillation/atrial flutter was reported in patients treated with Calquence+G and Calquence monotherapy with an incidence of 3% and 4%, respectively, including 1% with ≥ Grade 3 atrial fibrillation/atrial flutter in the Calquence+G arm. No patients experienced ≥ Grade 3 atrial fibrillation/atrial flutter in the Calquence monotherapy arm.
Infusion related reaction in ELEVATE-TN.
Infusion related reaction was reported in 14% and 40% of patients in the Calquence+G and GClb arms, respectively.
ASCEND (acalabrutinib monotherapy in relapsed/refractory CLL after at least one prior therapy). The safety of Calquence versus investigator's choice of either idelalisib plus rituximab or bendamustine plus rituximab was evaluated in a randomised, multicentre, open-label, Phase 3 study, in 307 patients with relapsed or refractory CLL. Details of the study treatment are described in Section 5.1 Pharmacodynamic Properties, Clinical trials.
In the Calquence arm, adverse events led to discontinuation in 10% and dose reduction in 3% of patients. In patients receiving idelalisib plus rituximab, adverse events led to regimen discontinuation in 9% of patients and a dose reduction of idelalisib in 24%. In patients receiving bendamustine plus rituximab, adverse events led to regimen discontinuation in 9% of patients and a dose reduction of bendamustine in 14% of patients. There were no dose reductions of rituximab.
The adverse reactions described in Table 11 and Table 12 reflect exposure to Calquence with a median duration of 15.7 months, exposure to idelalisib with a median duration of 11.5 months, exposure to rituximab with a median duration of 5.5 months, and exposure to bendamustine and a median duration of 5.6 months in patients with relapsed or refractory CLL.


Tumour lysis syndrome in ASCEND.
Tumour lysis syndrome (TLS) was reported in patients treated with Calquence and idelalisib plus rituximab with an incidence of 1% in both arms. The one patient experiencing TLS treated with Calquence had Grade 3 TLS and bulky disease.
Post-marketing experience.
The following adverse reactions have been identified during post-approval use of Calquence. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Vascular disorders.
Hypertension.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no specific treatment for acalabrutinib overdose and symptoms of overdose have not been established. In the event of an overdose, patients must be closely monitored for signs or symptoms of adverse reactions and appropriate symptomatic treatment instituted.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Acalabrutinib is a small-molecule inhibitor of Bruton's tyrosine kinase (BTK). Acalabrutinib and its active metabolite, ACP-5862, form a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. BTK is a signalling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. In B-cells, BTK signalling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis, and adhesion. In nonclinical studies, acalabrutinib inhibited BTK-mediated activation of downstream signalling proteins CD86 and CD69 and inhibited malignant B-cell proliferation and tumour growth in mouse xenograft models.
Pharmacodynamics.
In patients with B-cell malignancies dosed with 100 mg twice daily, median steady state BTK occupancy of ≥ 95% in peripheral blood was maintained over 12 hours, resulting in inactivation of BTK throughout the recommended dosing interval.
Cardiac electrophysiology.
The effect of acalabrutinib on the QTc interval was evaluated in a randomised, double-blind, double-dummy, placebo- and positive-controlled, 4-way crossover thorough QTc study in 48 healthy adult subjects. Administration of a single dose of acalabrutinib that is the 4-fold maximum recommended single dose did not prolong the QTc interval to any clinically relevant extent (i.e. ≥ 10 ms).
Clinical trials.
Mantle cell lymphoma (MCL). Patients with previously untreated MCL - ECHO.
The safety and efficacy of Calquence in patients with previously untreated MCL was evaluated in ECHO, a randomised, double-blind, placebo controlled, multicentre phase 3 study. ECHO enrolled 598 patients who were at least 65 years of age and had no intention to undergo autologous haematopoietic stem cell transplant. The study excluded patients with total bilirubin > 1.5 x upper limit of normal (ULN), AST or ALT > 2.5 x ULN, or estimated creatinine clearance of ≤ 50 mL/min. Randomisation was stratified by geographic region and simplified Mantle Cell Lymphoma International Prognostic Index (MIPI) score. The study enrolled patients during the COVID-19 pandemic.
Patients were randomised in a 1:1 ratio to receive either Calquence plus bendamustine and rituximab (Calquence + BR) or placebo + BR. Treatment in both arms was continued until disease progression or unacceptable toxicity, and was administered in 28-day cycles as follows:
Induction (maximum 6 cycles):
Calquence 100 mg (or matching placebo) orally twice daily, every day of the cycle.
Bendamustine 90 mg/m2 administered IV over 30 minutes on Days 1 and 2 of each cycle.
Rituximab 375 mg/m2 administered IV on Day 1 of each cycle.
Maintenance - for patients remaining in response (PR or CR) after induction:
Calquence 100 mg (or matching placebo) orally twice daily, every day of the cycle
Rituximab 375 mg/m2 administered IV on Day 1 of every other cycle; ceased after a maximum of 12 additional doses of rituximab (up to Cycle 30).
After discontinuation of rituximab, patients continued Calquence 100 mg (or matching placebo) as monotherapy, taken orally twice daily until disease progression or unacceptable toxicity.
Crossover to Calquence monotherapy was permitted for patients in the placebo plus BR arm at disease progression.
The median age was 71 years (range 65-86), 71% were male, 78% were Caucasian, and 93% had an ECOG performance status of 0-1. The simplified MIPI score was low (0-3) in 33%, intermediate (4-5) in 43% and high (6-11) in 24% of patients. A total of 38% of patients had tumour bulk ≥ 5 cm and 86% had Ann Arbor stage IV disease. Most (80%) had classic MCL histology, whilst 7.7% had blastoid and 5.5% had pleomorphic forms. A total of 48% patients had Ki-67 score of ≥ 30%. The baseline characteristics were similar between both treatment arms.
The primary endpoint was progression-free survival (PFS) as assessed by an Independent Review Committee (IRC) per Lugano Classification for NHL in subjects with previously untreated MCL. Additional efficacy endpoints were Investigator-assessed (INV) PFS, INV and IRC assessed overall response rate (ORR), IRC and INV assessed duration of response (DOR) and overall survival (OS).
Efficacy results are presented in Table 13 and Figure 1. At this prespecified interim analysis, the median follow-up for PFS was 49.8 months in both arms. Median OS had not been reached in any arm with a total of 203 deaths: 97 (32.4%) in the Calquence + BR arm, 106 (35.5%) in the Placebo + BR arm.


Patients with MCL who received at least one prior therapy - ACE-LY-004.
The safety and efficacy of Calquence in MCL were evaluated in an open-label, multi-centre, single-arm Phase 2 study (ACE-LY-004) of 124 previously treated patients. All patients received Calquence 100 mg orally twice daily until disease progression or unacceptable toxicity. The trial did not include patients who received prior treatment with BTK inhibitors. The primary endpoint was investigator-assessed overall response rate (ORR) per the Lugano classification for non-Hodgkin's lymphoma (NHL). Duration of Response (DoR) was an additional outcome measure.
The median age was 68 (range 42 to 90) years, 80% were male and 74% were Caucasian. At baseline, 93% of patients had an ECOG performance status of 0 or 1. The median time since diagnosis was 46 months and the median number of prior treatments was 2 (range 1 to 5), including 17.7% with prior stem cell transplant. The most common prior regimens were CHOP-based (52%) and ARA-C (34%). At baseline, 37% of patients had at least one tumour with a longest diameter ≥ 5 cm, 73% had extra nodal involvement including 51% with bone marrow involvement. The simplified MIPI score (which includes age, ECOG score, and baseline lactate dehydrogenase and white cell count) was intermediate in 44% and high in 17% of patients. The median dose intensity was 98.5%.
Efficacy results are presented in Table 14 for the primary analysis, and at a 54-month final analysis (median follow-up 38.1 months).
 Lymphocytosis. Upon initiation of Calquence, a temporary increase in lymphocyte counts (defined as absolute lymphocyte count (ALC) increased ≥ 50% from baseline and a post baseline assessment ≥ 5 x 109) in 32% of patients in ACE-LY-004. The median time to onset of lymphocytosis was 1.1 weeks and the median duration of lymphocytosis was 6.7 weeks.
Lymphocytosis. Upon initiation of Calquence, a temporary increase in lymphocyte counts (defined as absolute lymphocyte count (ALC) increased ≥ 50% from baseline and a post baseline assessment ≥ 5 x 109) in 32% of patients in ACE-LY-004. The median time to onset of lymphocytosis was 1.1 weeks and the median duration of lymphocytosis was 6.7 weeks.
Chronic lymphocytic leukaemia (CLL). Patients with previously untreated CLL- ELEVATE-TN.
The safety and efficacy of Calquence in previously untreated CLL were evaluated in a randomised, multi-centre, open-label Phase 3 study (ELEVATE-TN) of 535 patients. Patients received Calquence plus obinutuzumab, Calquence monotherapy, or obinutuzumab plus chlorambucil. Patients 65 years of age or older or between 18 and 65 years of age with coexisting medical conditions were included in ELEVATE-TN. The trial also allowed patients to receive antithrombotic agents other than warfarin or equivalent vitamin K antagonists.
Patients were randomised in a 1:1:1 ratio into 3 arms to receive:
Calquence plus obinutuzumab (Calquence+G): Calquence 100 mg was administered twice daily starting on Cycle 1 Day 1 until disease progression or unacceptable toxicity. Obinutuzumab was administered starting on Cycle 2 Day 1 for a maximum of 6 treatment cycles. Obinutuzumab 1000 mg was administered on Days 1 and 2 (100 mg on Day 1 and 900 mg on Day 2), 8 and 15 of Cycle 2 followed by 1000 mg on Day 1 of Cycles 3 up to 7. Each cycle was 28 days.
Calquence monotherapy: Calquence 100 mg was administered twice daily until disease progression or unacceptable toxicity.
Obinutuzumab plus chlorambucil (GClb): obinutuzumab and chlorambucil were administered for a maximum of 6 treatment cycles. Obinutuzumab 1000 mg was administered on Days 1 and 2 (100 mg on Day 1 and 900 mg on Day 2), 8 and 15 of Cycle 1 followed by 1000 mg on Day 1 of Cycles 2 up to 6. Chlorambucil 0.5 mg/kg was administered on Days 1 and 15 of Cycles 1 up to 6. Each cycle was 28 days.
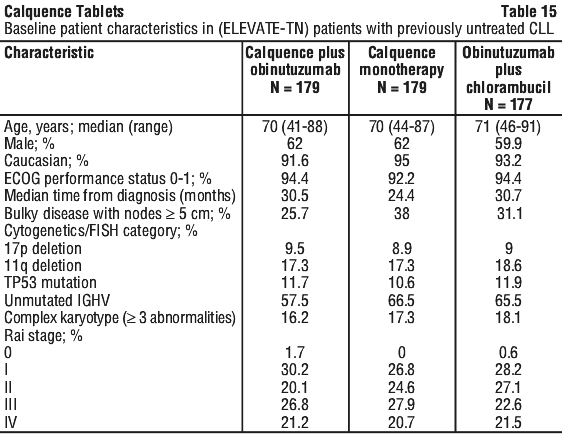
Patients were stratified by 17p deletion mutation status (presence versus absence), ECOG performance status (0 or 1 versus 2) and geographic region (North America and Western Europe versus Other). After confirmed disease progression, 45 patients randomised on the GClb arm crossed over to Calquence monotherapy. Table 15 summarises the baseline demographics and disease characteristics of the study population.
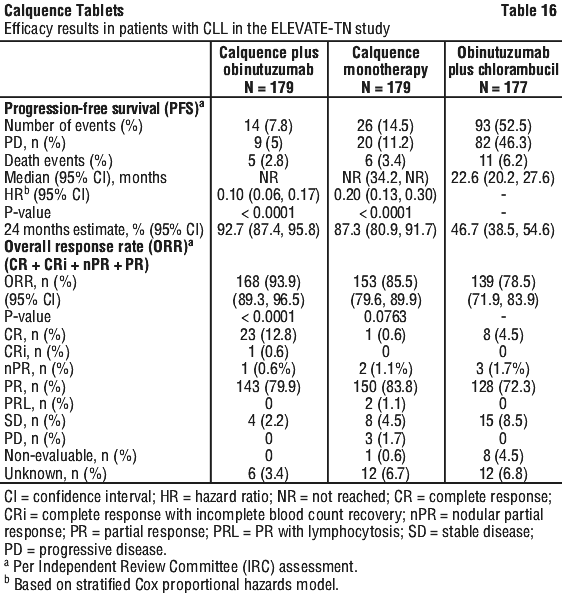
 The primary endpoint was progression-free survival (PFS) of Calquence+G arm versus GClb arm as assessed by an Independent Review Committee (IRC) per International Workshop on Chronic Lymphocytic Leukaemia (IWCLL) 2008 criteria with incorporation of the clarification for treatment-related lymphocytosis (Cheson 2012). With a median follow-up of 28.3 months, PFS by IRC indicated a 90% statistically significant reduction in the risk of disease progression or death for previously untreated CLL patients in the Calquence+G arm compared to the GClb arm. At the time of analysis, median overall survival had not been reached in any arm with a total of 37 deaths: 9 (5%) in the Calquence+G arm, 11 (6.1%) in the Calquence monotherapy arm, and 17 (9.6%) in the GClb arm. Efficacy results are presented in Table 16. The Kaplan-Meier curves for PFS are shown in Figure 2.
The primary endpoint was progression-free survival (PFS) of Calquence+G arm versus GClb arm as assessed by an Independent Review Committee (IRC) per International Workshop on Chronic Lymphocytic Leukaemia (IWCLL) 2008 criteria with incorporation of the clarification for treatment-related lymphocytosis (Cheson 2012). With a median follow-up of 28.3 months, PFS by IRC indicated a 90% statistically significant reduction in the risk of disease progression or death for previously untreated CLL patients in the Calquence+G arm compared to the GClb arm. At the time of analysis, median overall survival had not been reached in any arm with a total of 37 deaths: 9 (5%) in the Calquence+G arm, 11 (6.1%) in the Calquence monotherapy arm, and 17 (9.6%) in the GClb arm. Efficacy results are presented in Table 16. The Kaplan-Meier curves for PFS are shown in Figure 2.
 PFS results for Calquence with or without obinutuzumab were consistent across subgroups, including high risk features. In the high risk CLL population (17p deletion, 11q deletion, TP53 mutation, and unmutated IGHV), the PFS HRs of Calquence with or without obinutuzumab versus obinutuzumab plus chlorambucil was 0.08 [95% CI (0.04, 0.15)] and 0.15 [95% CI (0.09, 0.25)], respectively.
PFS results for Calquence with or without obinutuzumab were consistent across subgroups, including high risk features. In the high risk CLL population (17p deletion, 11q deletion, TP53 mutation, and unmutated IGHV), the PFS HRs of Calquence with or without obinutuzumab versus obinutuzumab plus chlorambucil was 0.08 [95% CI (0.04, 0.15)] and 0.15 [95% CI (0.09, 0.25)], respectively.
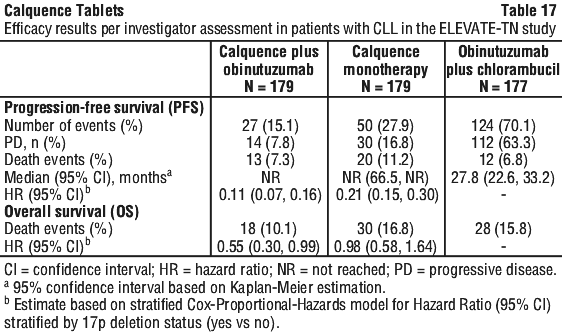
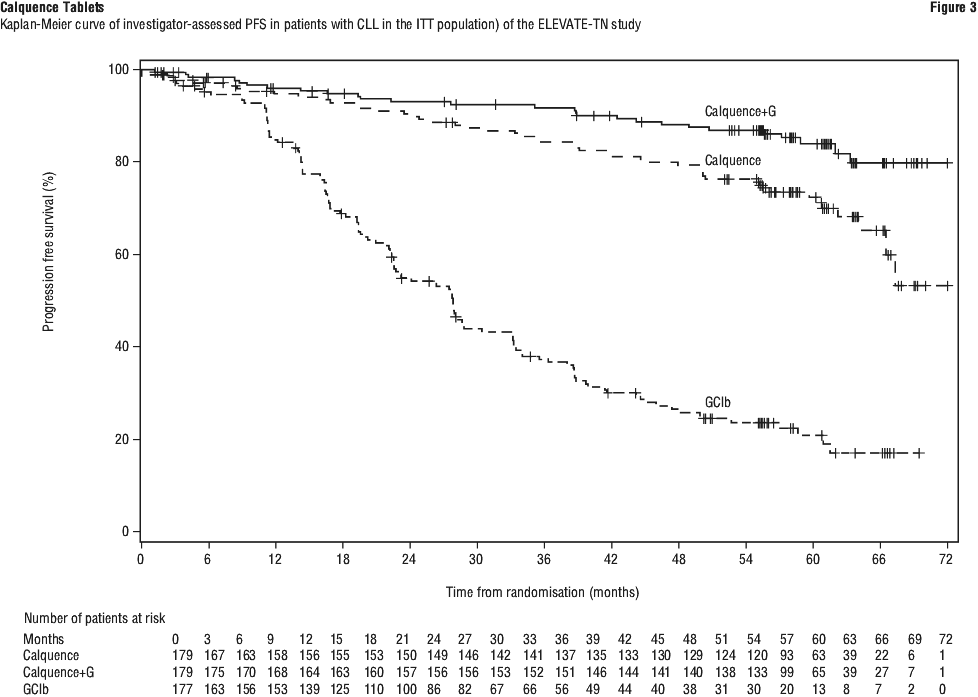
With long term data, the median follow-up was 58.2 months for Calquence+G arm, 58.1 months for Calquence arm and 58.2 months for the GClb arm. The median investigator-assessed PFS for Calquence+G and Calquence monotherapy was not reached; and was 27.8 months in GClb arm. At the time of most recent data cut off, a total of 72 patients (40.7%) originally randomised to the GClb arm crossed over to Calquence monotherapy. The median overall survival had not been reached in any arm with a total of 76 deaths: 18 (10.1%) in the Calquence+G arm, 30 (16.8%) in the Calquence monotherapy arm, and 28 (15.8%) in the GClb arm. The time to next treatment was prolonged for Calquence+G arm (HR=0.14 [95% CI: 0.09, 0.21]) and Calquence only arm (HR=0.27 [95% CI: 0.19, 0.38]) compared to GClb arm. See Table 17 and Figure 3.
Patients with CLL who received at least one prior therapy - ASCEND.
The safety and efficacy of Calquence in relapsed or refractory CLL were evaluated in a randomised, multi-centre, open-label Phase 3 study (ASCEND) of 310 patients who received at least one prior therapy. Patients received Calquence monotherapy or investigator's choice of either idelalisib plus rituximab or bendamustine plus rituximab. The trial allowed patients to receive antithrombotic agents other than warfarin or equivalent vitamin K antagonists.
Patients were randomised 1:1 to receive either:
Calquence 100 mg twice daily until disease progression or unacceptable toxicity, or
Investigator's choice: idelalisib 150 mg twice daily until disease progression or unacceptable toxicity in combination with ≤ 8 infusions of rituximab (375 mg/m2/500 mg/m2) on Day 1 of each 28-day cycle for up to 6 cycles.
Bendamustine 70 mg/m2 (Day 1 and 2 of each 28-day cycle) in combination with rituximab (375 mg/m2/500 mg/m2) on Day 1 of each 28-day cycle for up to 6 cycles.
Patients were stratified by 17p deletion mutation status (presence versus absence), ECOG performance status (0 or 1 versus 2) and number of prior therapies (1 to 3 versus ≥ 4). After confirmed disease progression, 35 patients randomised on investigator's choice of either idelalisib plus rituximab or bendamustine plus rituximab crossed over to Calquence. Table 18 summarises the baseline demographics and disease characteristics of the study population.
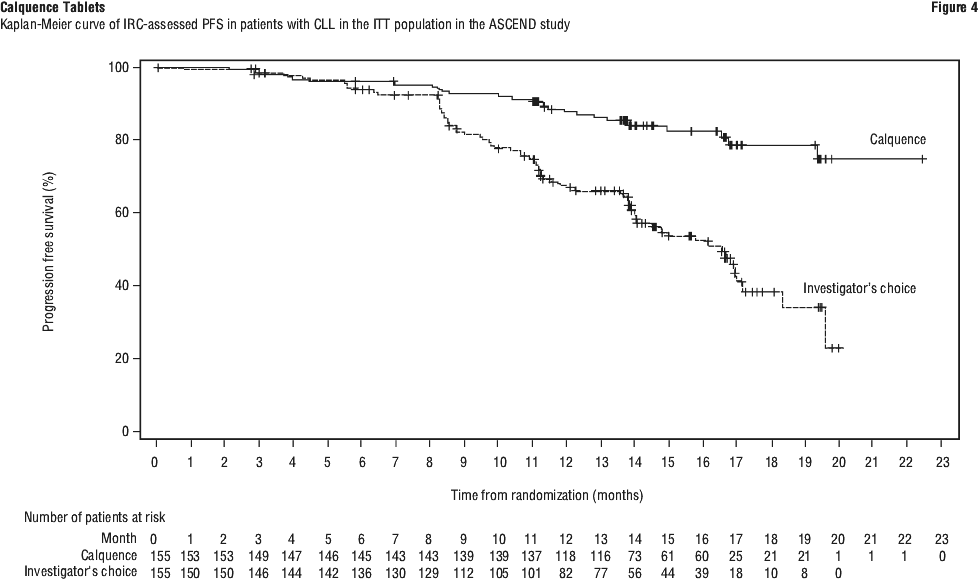
 The primary endpoint was PFS as assessed by IRC IWCLL 2008 criteria with incorporation of the clarification for treatment-related lymphocytosis (Cheson 2012). With a median follow-up of 16.1 months, PFS indicated a 69% statistically significant reduction in the risk of death or progression for patients in the Calquence arm. At the time of analysis, median overall survival had not been reached in any arm with a total of 33 deaths: 15 (9.7%) in the Calquence monotherapy arm and 18 (11.6%) in the investigator's choice of either idelalisib plus rituximab or bendamustine plus rituximab arm. Efficacy results are presented in Table 19. The Kaplan-Meier curve for PFS is shown in Figure 4.
The primary endpoint was PFS as assessed by IRC IWCLL 2008 criteria with incorporation of the clarification for treatment-related lymphocytosis (Cheson 2012). With a median follow-up of 16.1 months, PFS indicated a 69% statistically significant reduction in the risk of death or progression for patients in the Calquence arm. At the time of analysis, median overall survival had not been reached in any arm with a total of 33 deaths: 15 (9.7%) in the Calquence monotherapy arm and 18 (11.6%) in the investigator's choice of either idelalisib plus rituximab or bendamustine plus rituximab arm. Efficacy results are presented in Table 19. The Kaplan-Meier curve for PFS is shown in Figure 4.

 At the final analysis, with a median follow-up of 46.5 months for Calquence and 45.3 months for the IR/BR, 72% reduction in risk of investigator-assessed disease progression or death was observed for patients in the Calquence arm. The median investigator-assessed PFS was not reached in Calquence and was 16.8 months in IR/BR. The median investigator-assessed PFS in patients with 17p deletion per IWCLL criteria was not reached in Calquence arm and was 13.8 months in IR/BR arm.
At the final analysis, with a median follow-up of 46.5 months for Calquence and 45.3 months for the IR/BR, 72% reduction in risk of investigator-assessed disease progression or death was observed for patients in the Calquence arm. The median investigator-assessed PFS was not reached in Calquence and was 16.8 months in IR/BR. The median investigator-assessed PFS in patients with 17p deletion per IWCLL criteria was not reached in Calquence arm and was 13.8 months in IR/BR arm.
At the time of final analysis, median overall survival had not been reached in any arm with a total of 95 deaths: 41 (26.5%) in the Calquence monotherapy arm and 54 (34.8%) in the IR/BR arm. See Table 20 and Figure 5.
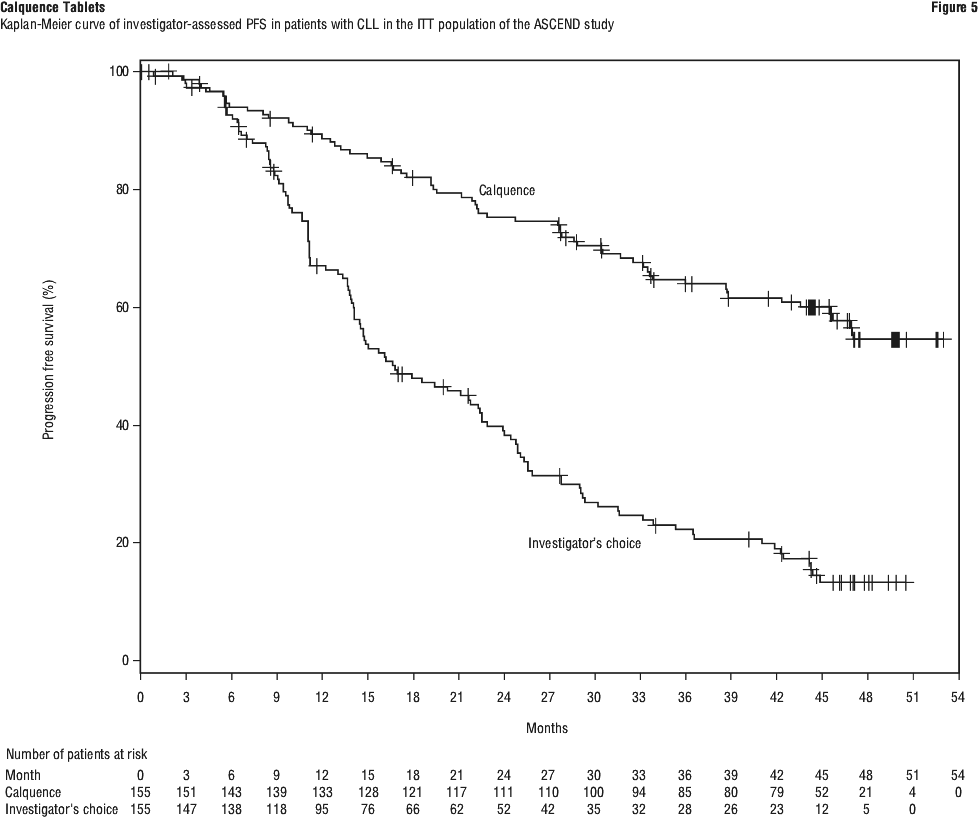 Investigator assessed PFS results for Calquence at final analysis were consistent across subgroups, including high risk features and were consistent with the primary analysis.
Investigator assessed PFS results for Calquence at final analysis were consistent across subgroups, including high risk features and were consistent with the primary analysis.
5.2 Pharmacokinetic Properties
The pharmacokinetics (PK) of acalabrutinib and its active metabolite, ACP-5862 were studied in healthy subjects and patients with B-cell malignancies. Acalabrutinib exhibits dose-proportionality, and both acalabrutinib and ACP-5862 exhibit almost linear PK across a dose range of 75 to 250 mg (0.75 to 2.5 times the approved recommended single dose). Population PK modelling suggests that the PK of acalabrutinib and ACP-5862 does not differ significantly in patients with different B-cell malignancies. At the recommended dose of 100 mg twice daily in patients with B-cell malignancies (including MCL and CLL), the geometric mean steady state daily area under the plasma drug concentration over time curve (AUC24h) and maximum plasma concentration (Cmax) of acalabrutinib were 1893 nanogram.h/mL and 466 nanogram/mL, respectively, and for ACP-5862 were 4091 nanogram.h/mL and 420 nanogram/mL, respectively.
Calquence tablets and Calquence capsules have been demonstrated to be bioequivalent. Geometric mean PK exposures (Cmax and AUClast or AUCinf) of acalabrutinib and ACP-5862 were similar (< 4% difference) between the tablet and capsule, with the 90% confidence intervals for geometric mean ratios within the pre-defined bioequivalence margin of 80% and 125%.
Absorption.
The median (min-max) time to peak plasma concentrations (Tmax) was 0.5 (0.2, 3.0) hours for acalabrutinib, and 0.75 (0.5, 4.0) hours for ACP-5862, following administration of acalabrutinib tablet. The absolute bioavailability of Calquence was 25% following oral administration of acalabrutinib capsule.
Distribution.
Reversible binding to human plasma protein was 97.5% for acalabrutinib and 98.6% for ACP-5862. The in vitro mean blood-to-plasma ratio was 0.8 for acalabrutinib and 0.7 for ACP-5862. The mean steady state volume of distribution (Vss) was approximately 34 L for acalabrutinib.
Metabolism.
In vitro, acalabrutinib is predominantly metabolised by CYP3A enzymes, and to a minor extent by glutathione conjugation and amide hydrolysis. ACP-5862 was identified as the major metabolite in plasma with a geometric mean exposure (AUC) that was approximately 2- to 3-fold higher than the exposure of acalabrutinib. ACP-5862 is approximately 50% less potent than acalabrutinib with regard to BTK inhibition.
Acalabrutinib may inhibit intestinal BCRP substrates (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions), while ACP-5862 may inhibit MATE1 (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions) at clinically relevant concentrations. Acalabrutinib does not inhibit MATE1, while ACP-5862 does not inhibit BCRP at clinically relevant concentrations.
Excretion.
Following a single oral dose of 100 mg acalabrutinib tablet, the median terminal elimination half-life (t1/2) of acalabrutinib was 1.3 (range: 0.8 to 9.0) hours. The median t1/2 of the active metabolite, ACP-5862, was 7.3 hours (range: 2.5 to 10.1) hours.
The mean apparent oral clearance (CL/F) was 70 L/hr for acalabrutinib and 13 L/hr for ACP-5862, with similar PK between patients and healthy subjects, based on population PK analysis.
Following administration of a single 100 mg radiolabelled [14C]-acalabrutinib dose in healthy subjects, 84% of the dose was recovered in the faeces and 12% of the dose was recovered in the urine, with less than 2% of the dose excreted as unchanged acalabrutinib in urine and faeces.
Specific populations.
Age, race, and body weight.
Age (32 to 90 years), sex, race (Caucasian, African American), and body weight (40 to 149 kg) did not have clinically meaningful effects on the PK of acalabrutinib and its active metabolite, ACP-5862, based on population PK analysis.
Renal impairment.
Acalabrutinib undergoes minimal renal elimination. Based on population PK analysis, no clinically relevant PK difference was observed in 543 patients with mild or moderate renal impairment (eGFR ≥ 30 mL/min/1.73 m2, as estimated by MDRD (modification of diet in renal disease equation)). Acalabrutinib PK has not been evaluated in patients with severe renal impairment (eGFR < 29 mL/min/1.73 m2, MDRD) or renal impairment requiring dialysis.
Hepatic impairment.
Acalabrutinib is metabolised in the liver. In hepatic impairment studies, compared to subjects with normal liver function (n = 6), acalabrutinib exposure (AUC) was increased by 1.9-fold, 1.5-fold, and 5.3-fold in subjects with mild (n = 6) (Child-Pugh A), moderate (n = 6) (Child-Pugh B) and severe (n = 8) (Child-Pugh C) hepatic impairment, respectively. Based on a population PK analysis, no clinically relevant PK difference was observed in subjects with mild (n = 79) or moderate (n = 6) hepatic impairment (total bilirubin between 1.5 to 3 times the upper limit of normal [ULN] and any AST) relative to subjects with normal (n = 651) hepatic function (total bilirubin and AST within ULN).
Drug interaction studies.
Effect of CYP3A inhibitors on acalabrutinib.
Co-administration with a strong CYP3A inhibitor (200 mg itraconazole once daily for 5 days) increased the acalabrutinib Cmax by 3.9-fold and AUC by 5.1-fold in healthy subjects.
Physiologically based pharmacokinetic (PBPK) simulations with acalabrutinib and moderate CYP3A inhibitors (erythromycin, fluconazole, diltiazem) showed that co-administration increased acalabrutinib Cmax and AUC increased by 2- to almost 3-fold (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Effect of CYP3A inducers on acalabrutinib.
Co-administration with a strong CYP3A inducer (600 mg rifampin once daily for 9 days) decreased acalabrutinib Cmax by 68% and AUC by 77% in healthy subjects (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Gastric acid reducing medicines.
Co-administration of acalabrutinib tablet with a proton pump inhibitor (20 mg rabeprazole, twice daily for 3 days) increased AUC by 17% (up to 31% increase, based on upper limit of 90% confidence interval) and decreased Cmax by 24% (up to 45% decrease, based on lower limit of 90% confidence interval), with a delay in Tmax (up to 1 hour, approximately).
5.3 Preclinical Safety Data
Genotoxicity.
Acalabrutinib was not mutagenic in an in vitro bacterial reverse mutation (AMES) assay or clastogenic in an in vitro human lymphocyte chromosomal aberration assay or in an in vivo rat bone marrow micronucleus assay.
Carcinogenicity.
Carcinogenicity studies have not been conducted with acalabrutinib.6 Pharmaceutical Particulars
6.1 List of Excipients
Tablet core.
Mannitol, microcrystalline cellulose, hyprolose, and sodium stearylfumarate.
Tablet coating.
Hypromellose, copovidone, titanium dioxide, macrogol 3350, medium chain triglycerides, iron oxide yellow, iron oxide red.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Polyamide-aluminium-polyvinylchloride/aluminium blisters. Cartons of 56 tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Acalabrutinib maleate monohydrate is a white to pale brown powder with pH-dependent solubility. It is freely soluble in water at pH values below 3 and practically insoluble at pH values above 6.
Chemical structure.
The chemical name is 4-{8-Amino-3-[(2S)-1-(but-2-ynoyl)pyrrolidin-2-yl]imidazo[1,5-a]pyrazin-1-yl}-N-(pyridin-2-yl)benzamide (2Z)-2-butenedioic acid hydrate (1:1:1).
 Molecular formula: C26H23N7O2.C4H4O4.H2O.
Molecular formula: C26H23N7O2.C4H4O4.H2O.
Molecular weight: 599.59.
CAS number.
CAS 2641500-53-8.7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Summary Table of Changes
