1 Name of Medicine
Mavacamten.
2 Qualitative and Quantitative Composition
Each 2.5 mg hard capsule contains 2.5 mg mavacamten.
Each 5 mg hard capsule contains 5 mg mavacamten.
Each 10 mg hard capsule contains 10 mg mavacamten.
Each 15 mg hard capsule contains 15 mg mavacamten.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Hard capsule (capsule).
Camzyos 2.5 mg capsules.
Light purple opaque cap imprinted with "2.5 mg" in black, and white opaque body imprinted with "Mava" in black, both in radial direction. Capsule size: 2.
Camzyos 5 mg capsules.
Yellow opaque cap imprinted with "5 mg" in black, and white opaque body imprinted with "Mava" in black, both in radial direction. Capsule size: 2.
Camzyos 10 mg capsules.
Pink opaque cap imprinted with "10 mg" in black, and white opaque body imprinted with "Mava" in black, both in radial direction. Capsule size: 2.
Camzyos 15 mg capsules.
Gray opaque cap imprinted with "15 mg" in black, and white opaque body imprinted with "Mava" in black, both in radial direction. Capsule size: 2.4.1 Therapeutic Indications
Camzyos is indicated for the treatment of adults with symptomatic NYHA class II-III obstructive hypertrophic cardiomyopathy.
4.2 Dose and Method of Administration
Treatment with mavacamten should be initiated and supervised by a specialist cardiologist, or consultant physician with experience in the management of obstructive hypertrophic cardiomyopathy (HCM).
Monitoring on treatment.
It is important to regularly monitor the patient's symptoms of obstructive HCM, left ventricular outflow tract (LVOT) gradient with Valsalva manoeuvre and left ventricular ejection fraction (LVEF) using echocardiogram assessments.
Follow the algorithms for Initiation of treatment (Figure 1) and Treatment maintenance (Figure 2) for appropriate Camzyos dosing and monitoring schedules. If at any visit the patient's LVEF is < 50%, the treatment should be interrupted for 4 weeks and until LVEF returns to ≥ 50%. Follow the algorithm for Interruption (Figure 3) for guidance on interrupting, restarting, or discontinuing Camzyos.
Based on the results of pharmacokinetic and exposure-response modelling and simulations, assessment of post-exercise LVOT gradient of > 30 mmHg may be considered in patients with normal or near normal Valsalva gradients (approximately 30 mmHg) as an additional echocardiogram parameter to guide dose changes after 12 weeks of Camzyos treatment.
Assessment of LVEF is recommended if clinical status changes or in patients with a serious intercurrent illness such as infection or arrhythmia (including atrial fibrillation or other uncontrolled tachyarrhythmia) (see Section 4.4 Special Warnings and Precautions for Use).
Testing prior to initiation with Camzyos.
Prior to initiating treatment with Camzyos, assess LVEF by echocardiography (see Section 4.4 Special Warnings and Precautions for Use). Treatment should not be initiated in patients with LVEF < 55%.
Recommended dosage.
The recommended starting dose of Camzyos is 5 mg orally once daily. The capsule should be swallowed whole with water and can be taken with or without food.
The patient's individualized daily dose of Camzyos will be either 2.5, 5, 10, or 15 mg. The maximum dose is 15 mg once daily.
Initiation of treatment.
The patient should be assessed for early clinical response 4 and 8 weeks after treatment initiation. See Figure 1.
If LVEF is < 50%, treatment should be interrupted.
Week 4 visit.
If LVOT gradient with Valsalva manoeuvre is < 20 mmHg, the dose should be decreased to 2.5 mg once daily. If LVOT gradient with Valsalva manoeuvre is ≥ 20 mmHg and LVEF remains ≥ 50%, maintain 5 mg once daily.
Week 8 visit.
If LVOT gradient with Valsalva manoeuvre is ≥ 20 mmHg and LVEF remains ≥ 50%, the dose of 2.5 mg or 5 mg once daily should be maintained. If LVOT gradient with Valsalva manoeuvre is < 20 mmHg, the dose should be decreased from 5 mg to 2.5 mg, and patients who are already on the lowest 2.5 mg dose should have their treatment paused until week 12.
All patients should return at week 12 for re-assessment. See Figure 1.

Treatment maintenance.
At Week 12, patients who paused treatment at week 8 should be reassessed, and if LVEF ≥ 50%, they can restart their treatment on 2.5 mg, regardless of LVOT gradient. A 4-week follow-up assessment visit is required to determine if the patient should maintain the 2.5 mg dose for the next 8 weeks. All other patients should follow the recommendations below. See Figure 2.
Week 12 and subsequent visits:
If LVEF is < 50%, regardless of LVOT gradient, treatment should be interrupted. Recheck echocardiogram parameters every 4 weeks until LVEF ≥ 50% to restart treatment at one lower dose level (e.g. 5 to 2.5 mg; 10 to 5 mg; 15 to 10 mg).
If LVEF is between 50% and < 55%, regardless of LVOT gradient, the current dose should be maintained. Assessment of LVEF and Valsalva LVOT gradient should be conducted every 3 months.
If LVEF is ≥ 55% and Valsalva LVOT gradient is < 30 mmHg, maintain the current dose. For patients in the first 6 months of treatment, recheck echocardiogram parameters in 3 months. If the results show LVEF ≥ 55% and Valsalva LVOT gradient < 30 mmHg and the patient has been on a stable dose for at least 6 months, echocardiogram monitoring can occur in 6 months.
For patients with LVEF ≥ 55% and Valsalva LVOT gradient ≥ 30 mmHg or post-exercise LVOT gradient ≥ 30 mmHg, if symptoms persist, the dose may be increased by one level (e.g. 2.5 to 5 mg; 5 to 10 mg; 10 to 15 mg).
A 4-week follow-up visit is required for any dose increase to determine if the patient should remain on the current dose for the next 8 weeks.
Monitor patients for symptoms of reduced LVEF. If symptoms occur, promptly evaluate cardiac function with an echocardiogram.
Dose increases are not recommended if the patient is experiencing an intercurrent illness such as infection or arrhythmia (including atrial fibrillation or other uncontrolled tachyarrhythmia) which may impair systolic function.

Treatment interruption if LVEF < 50%.
If at any clinical visit LVEF is < 50%, treatment should be interrupted. Restart treatment after 4 weeks at one lower dose level (e.g. 5 to 2.5 mg; 10 to 5 mg; 15 to 10 mg) if LVEF ≥ 50%. Patients on 2.5 mg who temporarily interrupt treatment due to LVEF < 50% on two consecutive occasions should discontinue treatment. Patients on 2.5 mg, whose LVEF is ≥ 50% and resumed treatment for four weeks after a temporary interruption due to LVEF < 50%, should discontinue treatment permanently if their LVEF is found to < 50%. at the next scheduled visit. See Figure 3.
 If during future 12-weekly assessment visits the patient's obstructive HCM symptoms have not improved and the LVOT gradient is ≥ 30 mmHg, a further dose increase by one level may be considered in patients with an LVEF ≥ 55% up to a maximum daily dose of 15 mg.
If during future 12-weekly assessment visits the patient's obstructive HCM symptoms have not improved and the LVOT gradient is ≥ 30 mmHg, a further dose increase by one level may be considered in patients with an LVEF ≥ 55% up to a maximum daily dose of 15 mg.
Dose increases should not occur more frequently than every 12 weeks. Following any dose increase, LVOT gradient with Valsalva manoeuvre and LVEF should be assessed after 4 weeks, and the patient should return 8 weeks later. Subsequent assessment of LVEF and Valsalva LVOT gradient may be done every 6 months, provided the patient has been on stable dose for 6 months.
Concomitant administration of weak to moderate CYP2C19 or moderate to strong CYP3A4 inhibitors.
Concomitant use with moderate CYP2C19 inhibitors or strong CYP3A4 inhibitors may be considered with appropriate dosing and caution.
Concomitant use with a moderate CYP2C19 inhibitor or a strong CYP3A4 inhibitor increases mavacamten exposure, which may increase the risk of adverse events.
It is recommended that patients who initiate or modify treatment with weak to moderate CYP2C19 inhibitors or moderate to strong CYP3A4 inhibitors, consider additional monitoring of LVEF. Additionally, based on modelling and simulation, dosing recommendations for concomitant administration have been provided below.
Concomitant use with both a moderate CYP2C19 inhibitor and a strong CYP3A4 inhibitor is not recommended.
Patients on moderate CYP2C19 inhibitors or strong CYP3A4 inhibitors.
In patients who are on stable therapy with a moderate CYP2C19 inhibitor or a strong CYP3A4 inhibitor, initiate Camzyos at 2.5 mg orally once daily. Camzyos should be stopped and coadministration should be avoided if LVEF < 50% occurs while receiving 2.5 mg of Camzyos with a moderate CYP2C19 inhibitor or a strong CYP3A4 inhibitor. Interrupt Camzyos treatment if Valsalva LVOT gradient is < 20 mmHg at Week 4 or Week 8. Treatment with Camzyos may be resumed after 4 weeks at 2.5 mg once daily if LVEF remains ≥ 50%. If treatment is resumed at Week 12, recheck clinical status, Valsalva LVOT gradient and LVEF in 4 weeks, and maintain the current dose for the next 8 weeks unless LVEF is < 50%.
Patients on weak CYP2C19 inhibitors or moderate CYP3A4 inhibitors.
Initiate Camzyos at the recommended starting dosage of 5 mg orally once daily in patients who are on stable therapy with a weak CYPC219 inhibitor or a moderate CYP3A4 inhibitor (see Figure 1).
Patients on mavacamten initiating weak to moderate CYP2C19 or moderate to strong CYP3A4 inhibitors.
In patients who initiate a weak to moderate CYP2C19 inhibitor or a moderate to strong CYP3A4 inhibitor, reduce dosage of CAMZYOS to the next lower daily (mg) dose level (i.e. 15 mg to 10 mg; 10 mg to 5 mg; or 5 mg to 2.5 mg). Schedule clinical and echocardiographic assessment 4 weeks after inhibitor initiation, and do not up-titrate to the next higher daily (mg) dose level of Camzyos until 12 weeks after inhibitor initiation. Avoid initiation of concomitant weak to moderate CYP2C19 or moderate to strong CYP3A4 inhibitors in patients who are on stable treatment with 2.5 mg of Camzyos because a lower Camzyos once-daily dose is not available (see Section 4.3 Contraindications; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Camzyos should be stopped and coadministration should be avoided if LVEF < 50% occurs while receiving 2.5 mg of Camzyos with a moderate CYP2C19 or a strong CYP3A4 inhibitor.
Short-term use of weak to moderate CYP2C19 inhibitors or moderate to strong CYP3A4 inhibitors.
For short-term use (e.g. 1 week), interrupt Camzyos for the duration of treatment with a weak to moderate inhibitor of CYP2C19 or a moderate to strong inhibitor of CYP3A4. Camzyos may be reinitiated at the previous dose immediately on discontinuation of concomitant therapy.
Missed or delayed doses.
If a dose is missed, it should be taken as soon as possible, and the next scheduled dose should be taken at the usual time the following day. Two doses should not be taken on the same day.
Renal impairment.
No dosage adjustment is needed in patients with mild (eGFR 60 to < 90 mL/min/1.73 m2) to moderate (eGFR 30 - < 60 mL/min/1.73 m2) renal impairment. Caution should be used in patients with severe (eGFR < 30 mL/min/1.73 m2) renal impairment, as Camzyos has not been studied in this population (see Section 5.2 Pharmacokinetic Properties, Special populations).
Hepatic impairment.
No dosage adjustment is required for patients with mild (Child-Pugh A) to moderate (Child-Pugh B) hepatic impairment. Caution should be used in patients with severe (Child-Pugh C) hepatic impairment, as Camzyos has not been studied in this population (see Section 5.2 Pharmacokinetic Properties, Special populations).
Paediatric and adolescent.
The safety and efficacy of Camzyos in paediatric patients aged less than 18 years of age have not been established. No data are available.
Elderly.
No dosage adjustment is required in patients 65 years and older (see Section 5.2 Pharmacokinetic Properties, Special populations).4.3 Contraindications
Hypersensitivity to mavacamten or to any of the excipients (see Section 6.1).
Concomitant use with:
Strong CYP2C19 inhibitors (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions);
moderate to strong CYP2C19 inducers or moderate to strong CYP3A4 inducers (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
4.4 Special Warnings and Precautions for Use
Heart failure with reduced ejection fraction.
Camzyos reduces LVEF and may cause heart failure with reduced ejection fraction (HFrEF) defined as symptomatic LVEF < 50%. Patients with a serious intercurrent illness such as serious infections or arrhythmia (including atrial fibrillation or other uncontrolled tachyarrhythmia) or those undergoing major cardiac surgery may be at greater risk of systolic dysfunction and progress to heart failure (see Section 4.8 Adverse Effects (Undesirable Effects)). New or worsening dyspnoea, chest pain, fatigue, palpitations, leg oedema or elevations in N-terminal-pro-B-type natriuretic peptide (NT-proBNP) may be signs and symptoms of a reduced LVEF and should prompt an evaluation of cardiac function. LVEF should be measured prior to initiating treatment and closely monitored thereafter. Treatment with Camzyos should not be initiated in patients with LVEF < 55%. Treatment interruption may be necessary to ensure that LVEF remains ≥ 50% (see Section 4.2 Dose and Method of Administration).
Risk of heart failure or loss of response to mavacamten due to drug-drug interactions.
Camzyos is primarily metabolised by cytochrome P450 (CYP) 2C19 and CYP 3A4 enzymes. Concomitant administration of Camzyos with CYP2C19 or CYP3A4 inhibitors or inducers may lead to increased risk of HFrEF or a loss of therapeutic response. As cessation of stable, chronic CYP2C19 and CYP3A4 inhibitors could potentially lead to a reduction in mavacamten exposure and efficacy, close clinical echocardiogram monitoring is advised in these situations (see Section 4.3 Contraindications; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions):
Prior to and during Camzyos treatment, the potential for drug interactions, including over-the-counter medications (such as omeprazole or esomeprazole), should be considered. Advise patients to inform their healthcare provider of all concomitant products prior to and during Camzyos treatment.
Concomitant use of negative inotropes.
Due to limited data, the safety of concomitant use of Camzyos with disopyramide, or use of Camzyos in patients taking beta blockers in combination with verapamil or diltiazem has not been established. Therefore, caution should be used when taking these concomitant medications and patients should be closely monitored (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Embryo-fetal toxicity.
Based on animal studies, mavacamten may cause embryo-fetal harm when administered to a pregnant woman. Women of childbearing potential and women becoming pregnant while receiving the treatment should be informed of the potential risk to the fetus. Women of childbearing potential have to use highly effective contraception during treatment with Camzyos and for at least 4 months after discontinuing treatment (see Section 4.6 Fertility, Pregnancy and Lactation).
Use in hepatic impairment.
Caution should be used in patients with severe (Child-Pugh C) hepatic impairment, as Camzyos has not been studied in this population (see Section 5.2 Pharmacokinetic Properties, Special populations).
Use in renal impairment.
Caution should be used in patients with severe (eGFR < 30 mL/min/1.73 m2) renal impairment, as Camzyos has not been studied in this population (see Section 5.2 Pharmacokinetic Properties, Special populations).
Use in the elderly.
Safety, effectiveness, and pharmacokinetics were consistent between elderly patients (≥ 65 years) and younger patients (18 to < 65 years) (see Section 5.2 Pharmacokinetic Properties, Special populations).
Paediatric use.
No data available.
Effects on laboratory tests.
The effects of Camzyos on laboratory tests were not considered clinically significant.4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of other drugs on Camzyos.
Camzyos is primarily metabolised by CYP2C19 and to a lesser extent by CYP3A4. Moderate to strong CYP3A4 inhibitors/ inducers or any CYP2C19 inhibitors/ inducers may thus affect the clearance of Camzyos and increase/decrease Camzyos plasma concentration (see Section 4.4 Special Warnings and Precautions for Use).
CYP 2C19 and CYP 3A4 inhibitors.
Coadministration of mavacamten with a weak CYP2C19 inhibitor (omeprazole) resulted in a 48% increase in mavacamten AUCinf with no effect on Cmax. Coadministration of mavacamten with a strong CYP3A4 inhibitor (itraconazole) resulted in an increase in mavacamten plasma concentration of up to 59% and 40% in AUC0-24 and Cmax, respectively.
Coadministration of mavacamten with a moderate CYP3A4 inhibitor (verapamil) resulted in an increase in mavacamten plasma concentration of 16% and 52% in AUCinf and Cmax, respectively.
Concomitant use with a strong CYP2C19 inhibitor is contraindicated (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use).
If initiating or adjusting the dose of a concomitant weak to moderate CYP2C19 inhibitor or moderate to strong CYP3A4 inhibitor, increased clinical assessments and dose adjustment are advised. Coadministration of mavacamten with both a moderate CYP2C19 inhibitor and a strong CYP3A4 inhibitor is not recommended (see Section 4.2 Dose and Method of Administration).
CYP 2C19 and CYP 3A4 inducers.
Coadministration of mavacamten with a strong CYP2C19 and CYP3A4 inducer following a 7-day lead-in induction period, is predicted by PBPK modelling to result in a decrease in mavacamten of up to 60% and 7% in AUC0-t and Cmax, respectively in CYP2C19 normal metabolisers (NM). In CYP2C19 poor metabolisers (PM) a decrease of up to 69% and 4% in AUC0-t and Cmax was predicted. Mavacamten clearance is expected to increase for both NM and PM by 2.5-fold and 3.2-fold, respectively, under induction conditions.
Concomitant use with a moderate or strong CYP2C19 inducer or a moderate or strong CYP3A4 inducer is contraindicated (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use).
Coadministration of mavacamten with a weak CYP2C19 or a weak CYP3A4 inducer may result in a decrease in mavacamten plasma concentration. If discontinuing concomitant treatment with a weak CYP2C19 or CYP3A4 inducer, monitor LVEF 4 weeks after inducer discontinuation, subsequently resume the monitoring and titration schedule (see Section 4.2 Dose and Method of Administration, Figure 1, Figure 2).
Effect of Camzyos on CYP3A4 substrates.
Coadministration of a 16-day course of mavacamten resulted in a decrease in midazolam plasma concentration. This change was not considered clinically significant. Coadministration of a 17-day course of mavacamten did not decrease the exposure to ethinyl oestradiol and norethindrone, which are the components of typical oral contraceptives and substrates for CYP3A4.
Effect of mavacamten on other CYP substrates.
Based on pre-clinical data, mavacamten is not an inhibitor of CYP 1A2, 2B6, 2C8, 2D6, 2C9, 2C19, or 3A4 at clinically relevant concentrations.
In vitro, mavacamten is an inducer of CYP 2B6, and 3A4, 2C8, 2C19 and 2C9.
Effect of mavacamten on transporters.
In vitro data indicate that mavacamten is not an inhibitor of major efflux transporters (P-gp, BCRP, BSEP, MATE1, or MATE2-K) or major uptake transporters (organic anion transporting polypeptides [OATPs], organic cation transporters [OCTs], or organic anion transporters [OATs]) at therapeutic concentrations.
Other interactions.
Drugs that reduce cardiac contractility.
In the EXPLORER-HCM study, 119 of 123 patients who received Camzyos received concomitant treatment with either beta blockers, verapamil, or diltiazem (see Section 5.1 Pharmacodynamic Properties, Clinical trials). In the VALOR-HCM study, 53 of the 56 patients who received Camzyos during the randomised controlled period received concomitant therapy with the following medications (alone or in combination with other treatment): beta blocker, verapamil or diltiazem, and/or disopyramide. In the overall study, including patients who were treated with Camzyos after the double-blind period, 36 of 112 patients (32%) received Camzyos with combination background HCM therapy; 22 of 112 patients (20%) received disopyramide as monotherapy or in combination with other treatments and no evidence of systolic dysfunction was observed. There is limited information available on the potential for a pharmacodynamic (PD) interaction between Camzyos and other drugs that also reduce cardiac contractility. If treatment with a new negative inotrope is initiated, or if the dose of a negative inotrope is increased, in a patient receiving Camzyos, close medical supervision with echocardiographic monitoring of LVEF should be provided until stable doses and clinical response have been achieved (see Section 4.4 Special Warnings and Precautions for Use).4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
In reproductive toxicity studies, there was no evidence of effects of mavacamten on mating and fertility in male or female rats or on the viability and fertility of offspring of dams at any dose tested. Plasma exposures (AUC) of mavacamten at the highest doses tested are less than in humans at the maximum recorded human dose (MRHD).
(Category D)
There are no adequate data on the developmental risk associated with the use of Camzyos in pregnant females. Based on animal data, Camzyos may cause fetal harm when administered to a pregnant female.
Camzyos should not be administered to patients who are pregnant. Females of reproductive potential who undergo treatment with Camzyos should be informed of the potential hazard to the foetus and should be advised to avoid becoming pregnant prior to or during treatment and for at least 4 months after discontinuation.
If the patient becomes pregnant while receiving the drug, the patient should be informed of the potential hazard to the foetus.
Pregnancy testing.
Confirm a negative pregnancy test in women of reproductive potential prior to initiation of treatment.
Contraception.
Advise females of reproductive potential to avoid becoming pregnant and to use highly effective contraception during treatment with Camzyos and for at least 4 months after discontinuing treatment.
Animal data.
When mavacamten was administered orally to pregnant rats during the period of organogenesis, decreased mean fetal body weight, and increases in post implantation loss and fetal malformations (visceral and skeletal) were observed in the high dose group (1.5 mg/kg/day). Visceral malformations (heart malformation in foetuses, including one total situs inversus) and increased incidences of skeletal malformations (mainly fused sternebrae) were observed. Plasma exposure (AUC) at the no effect dose for embryo-fetal development in rats and rabbits are less than those in humans at the MRHD.
When mavacamten was administered orally to pregnant rabbits during the period of organogenesis, fetal malformations (external, visceral and skeletal) were increased at doses of 1.2 mg/kg/day and higher. Visceral findings consisted of malformations of the great vessels (pulmonary trunk atresia, dilatation of pulmonary trunk) and malformations of the heart, kidney, ureter and testis at 2.0 mg/kg/day. Skeletal malformations consisted of higher incidences of fused sternebrae at 2.0 mg/kg/day. External findings consisted of cleft palate at doses of 1.2 mg/kg/day and higher. Plasma exposure (AUC) at the no effect dose for embryo-fetal development in rabbits is less than those in humans at the MRHD.
It is unknown whether mavacamten or its metabolites are excreted in human milk. Because of the unknown adverse effects of mavacamten in breastfed newborns/infants, a decision must be made whether to discontinue breastfeeding during treatment and for 4 months after the last dose or to discontinue treatment, taking into account the benefit of breastfeeding for the child and the benefit of treatment for the woman.
In a pre- and post-natal development study, mavacamten was administered orally to pregnant rats from gestation day 6 to lactation/post-partum day 20. No adverse effects were observed in the dams or offspring exposed daily from before birth (in utero) through lactation. The maternal exposure was inferred from the embryo-fetal developmental toxicity study dosed at the same level, and the exposure was less than the MRHD.4.7 Effects on Ability to Drive and Use Machines
Camzyos may have minor influence on the ability to drive and use machines. Dizziness may occur following administration of mavacamten. Patients should be advised not to drive or use machines if they experience dizziness.
4.8 Adverse Effects (Undesirable Effects)
Clinical experience.
The safety of Camzyos was evaluated in EXPLORER-HCM, a Phase 3, double-blind, randomized, placebo controlled, trial. Of the 251 obstructive HCM adult patients in this trial, 123 patients were treated with a daily dose of either 2.5 mg, 5 mg, 10 mg or 15 mg of Camzyos and 128 were treated with placebo. Camzyos-treated patients received a median duration of exposure of 30.4 weeks (range: 1.6 to 40.3 weeks).
There were no adverse reactions leading to discontinuation. Two patients out of 123 (1.6%) in the Camzyos group and no patients (0%) in the placebo group discontinued trial drug. In the Camzyos group, the adverse events leading to discontinuation were syncope (0.8%) and atrial fibrillation (0.8%) in one patient each.
Adverse events reported by ≥ 5% of subjects in any treatment group are provided in Table 1.
 The adverse drug reactions (ADRs) according to system organ class in MedDRA are listed in Table 2. Within each system organ class, the ADRs are presented in order of decreasing seriousness. In addition, the corresponding frequency category for each ADR is defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000).
The adverse drug reactions (ADRs) according to system organ class in MedDRA are listed in Table 2. Within each system organ class, the ADRs are presented in order of decreasing seriousness. In addition, the corresponding frequency category for each ADR is defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000).
 The safety of Camzyos in patients was further evaluated in VALOR-HCM, a phase 3, double-blind, randomized, placebo-controlled trial. Of the 112 adults with symptomatic obstructive HCM, 56 patients were treated with Camzyos 2.5-15 mg daily and 55 were treated with placebo. Camzyos treated patients had a median duration of exposure of 17 weeks (range: 3-19 weeks) during the double-blind period (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
The safety of Camzyos in patients was further evaluated in VALOR-HCM, a phase 3, double-blind, randomized, placebo-controlled trial. Of the 112 adults with symptomatic obstructive HCM, 56 patients were treated with Camzyos 2.5-15 mg daily and 55 were treated with placebo. Camzyos treated patients had a median duration of exposure of 17 weeks (range: 3-19 weeks) during the double-blind period (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
There were no adverse drug reactions leading to discontinuation in patients receiving Camzyos. During the placebo-controlled period of the study, adverse reaction occurring in > 5% of patients and more commonly on Camzyos than on placebo was dizziness (13% vs. 6%).
Adverse events reported by ≥ 5% of subjects in any treatment group during placebo-controlled period of VALOR-HCM trial are provided in Table 3.

Effects on systolic function.
In the EXPLORER-HCM trial, 7 (6%) patients in the Camzyos group and 2 (2%) patients in the placebo group experienced reversible reductions in LVEF < 50% (median 48%: range 35-49%) while on treatment. None of the 7 patients receiving mavacamten had systolic dysfunction leading to heart failure. In 3 of the 7 Camzyos patients and in 1 of the 2 placebo patients, these reductions were observed without other clinical manifestations (e.g. symptoms). In all 7 patients treated with Camzyos, LVEF recovered following interruption of Camzyos and they completed the study (see Section 4.4 Special Warnings and Precautions for Use).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.4.9 Overdose
Human experience of overdose with Camzyos is limited. Camzyos has been given as a single dose of up to 144 mg in patients with HCM. There was one serious adverse reaction of vasovagal reaction, hypotension, and asystole lasting 38 seconds reported at that dose. In healthy subjects, doses of up to 25 mg have been administered for up to 25 days. Three out of 8 participants treated at the 25 mg dose level experienced 20% or greater reductions in LVEF. Systolic dysfunction is the most likely result of overdosage of Camzyos.
Management of overdose.
If warranted, treatment of overdose with Camzyos consists of discontinuation of Camzyos treatment as well as medically supportive measures to maintain hemodynamic status (e.g. initiation of inotropic support with adrenergic agents), including close monitoring of vital signs and LVEF and management of the clinical status of the patient.
In an open-label randomised pharmacokinetic (PK) study in healthy subjects fasted overnight, administration of activated charcoal 2 hours (approximately Tmax) after ingestion of a 15 mg dose of mavacamten reduced AUC and half-life of mavacamten, with a geometric mean ratio of 0.658 and 0.648, respectively. Administration of activated charcoal 6 hours after the mavacamten dose had minimal effect on mavacamten exposure and elimination. Thus, early administration (prior to or as soon after Tmax as possible) of activated charcoal may be considered in the management of mavacamten overdose or accidental ingestion. Under fed conditions, activated charcoal may still be effective beyond 2-hour post mavacamten dose because of the delayed Tmax by food (see Section 5.2 Pharmacokinetic Properties).
In patients who are not fully conscious or have impaired gag reflex, consideration should be given to administering activated charcoal via a nasogastric tube, once the airway is protected.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Mavacamten is a selective, allosteric, and reversible cardiac myosin inhibitor. Mavacamten modulates the number of myosin heads that can enter power-generating states, thus reducing, (or in HCM normalizing), the probability of force-producing systolic and residual diastolic crossbridge formation. Mavacamten also shifts the overall myosin population towards an energy sparing, but recruitable, super-relaxed state. Excess cross-bridge formation and dysregulation of the super-relaxed state of myosin are mechanistic hallmarks of HCM, which can result in hypercontractility, impaired relaxation, excess energy consumption, and myocardial wall stress. In HCM patients, myosin inhibition with mavacamten normalizes contractility, reduces dynamic LVOT obstruction, and improves cardiac filling pressures and biomarkers of cardiac stress, improving symptoms and exercise capacity.
LVEF.
A reduction in ejection fraction is expected with mavacamten treatment. In the EXPLORER-HCM trial, mean (SD) resting LVEF was 74% (6) at baseline in both treatment groups. Consistent with the mechanism of action of Camzyos, reductions in mean (SD) absolute change from baseline in LVEF was -4% (8) in the Camzyos group and 0% (7) in the placebo group over the 30-week treatment period. At Week 38, following an 8-week interruption of trial drug, mean LVEF was similar to baseline for both treatment groups.
LVOT obstruction.
In the EXPLORER-HCM trial, patients achieved significant reductions in mean resting and provoked (Valsalva) LVOT gradient by Week 4 which were sustained throughout the 30-week trial. At Week 30, the mean (SD) change from baseline in resting and Valsalva LVOT gradients were 39 (29) mmHg and -49 (34) mmHg, respectively, for the Camzyos group and -6 (28) mmHg and -12 (31) mmHg, respectively, for the placebo group. The clinically significant reductions in LVOT gradient were mediated by the mechanism of action of mavacamten, which resulted in small decreases in LVEF, attenuating the hypercontractility typical of obstructive HCM while maintaining LVEF within the normal range. The reductions in LVOT gradient while maintaining stable heart rate and systemic blood pressure demonstrate improvement in forward blood flow. At Week 38, following 8 weeks of trial drug washout, mean LVEF and LVOT gradients were similar to baseline for both treatment groups.
Cardiac structure.
In the EXPLORER-HCM trial, echocardiographic measurements of cardiac structure showed a mean (SD) reduction from baseline at Week 30 in left ventricular mass index (LVMI) in the mavacamten group of -7.4 (17.8) g/m2 versus an increase in LVMI in the placebo group of 9.0 (15.3) g/m2. There was also a mean (SD) reduction from baseline in left atrial volume index (LAVI) in the mavacamten group -7.5 (7.8) mL/m2 versus no change in the placebo group with -0.1 (8.7) mL/m2.
Cardiac electrophysiology.
In HCM, the QT interval may be intrinsically prolonged due to the underlying disease, in association with ventricular pacing, or in association with drugs with potential for QT prolongation commonly used in HCM population. An exposure-response analysis across all clinical studies in HCM patients has shown a concentration-dependent shortening of the QTcF interval. The mean placebo corrected change from baseline in obstructive HCM patients was -8.7 ms (upper and lower limit of the 90% CI -6.7 ms and -10.8 ms, respectively) at the median steady-state Cmax of 452 nanogram/mL. Patients with longer baseline QTcF intervals tended to display the greatest shortening. In the EXPLORER-HCM trial, there was no evidence of an increase in clinical events suggestive of ventricular arrhythmias (e.g. sudden deaths, syncope, or seizures) in the mavacamten group compared to placebo. There is limited experience on co-administration of mavacamten with QT prolonging drugs or in patients with potassium channel variants resulting in a long QT interval. In the VALOR-HCM trial, addition of mavacamten did not appear to worsen the prolongation of QTcF that can be present with disopyramide treatment.
Consistent with nonclinical findings in normal hearts, in one clinical trial in healthy subjects sustained exposure to mavacamten at supratherapeutic levels leading to marked depression of systolic function was associated with QTc prolongation (< 20 ms). No acute QTc changes have been observed at comparable (or higher) exposures after single doses.
Preclinical studies to investigate the observed QTc prolongation in healthy hearts in animals demonstrated no proarrhythmic and/or torsadogenic potential either in vivo, in vitro, and/or in silico, and confirmed that the QTc prolongation observed in healthy hearts is not the result of an off-target direct effect of mavacamten on late-repolarization currents like hERG ion channel activity and/or trafficking. The findings in healthy hearts are attributed to an adaptive response to the cardiac mechanical/functional changes (marked mechanical LV depression) occurring in response to myosin inhibition in hearts with normal physiology and LV contractility.
Cardiac biomarkers.
In the EXPLORER-HCM trial, reductions in a biomarker of cardiac wall stress, N-terminal pro-B-type natriuretic peptide (NT-proBNP) were observed by Week 4, and sustained through the end of treatment. The Week 30 ratio to baseline geometric mean was 0.20 for mavacamten and 1.02 for placebo (proportion of geometric mean ratio between the two arms 0.20, [95% CI: 0.17, 0.24]); therefore, the reduction in NT-proBNP after mavacamten treatment was 80% greater than for placebo.
Clinical trials.
Clinical efficacy and safety. EXPLORER-HCM trial.
The efficacy of Camzyos was evaluated in a double-blind, randomised, placebo-controlled, parallel-arm, multicentre, international, Phase 3 study (EXPLORER-HCM) enrolling 251 adult patients with symptomatic NYHA class II and III obstructive HCM, LVEF ≥ 55%, and LVOT peak gradient ≥ 50 mmHg at rest or with provocation. The majority of patients received background HCM treatment for a total of 96% in Camzyos arm (beta blockers 76%, calcium channel blockers 20%) and of 87% in the placebo arm (beta blockers 74%, calcium channel blockers 13%).
Patients were randomised in a 1:1 ratio to receive either a starting dose of 5 mg of Camzyos (123 patients) or matching placebo (128 patients) once daily for 30 weeks. The dose was periodically adjusted based on plasma concentrations of Camzyos and pharmacodynamic response (decrease in LVOT gradient with Valsalva manoeuvre and maintenance of LVEF ≥ 50%) to optimise patients' response. Within the dose range of 2.5 mg to 15 mg, a total of 81% (100/123) of patients were receiving either the 5 mg or 10 mg dose at the end of the treatment period, with 49% (60/123) receiving the 5 mg dose. During the study, 3 of 7 patients on Camzyos had LVEF < 50% prior to the Week 30 visit and temporarily interrupted their dose; 2 patients resumed treatment at the same dose and 1 patient had the dose reduced from 10 mg to 5 mg.
Treatment assignment was stratified by baseline disease severity NYHA functional class (II or III), current treatment with beta blockers (yes or no), type of ergometer (treadmill or exercise bicycle) used for assessment of peak oxygen consumption (pVO2). Patients on background dual treatment with beta blocker and calcium channel blocker treatment or disopyramide or ranolazine were excluded. Patients with known infiltrative or storage disorder causing cardiac hypertrophy that mimicked obstructive HCM, such as Fabry disease, amyloidosis, or Noonan syndrome with LV hypertrophy, were also excluded.
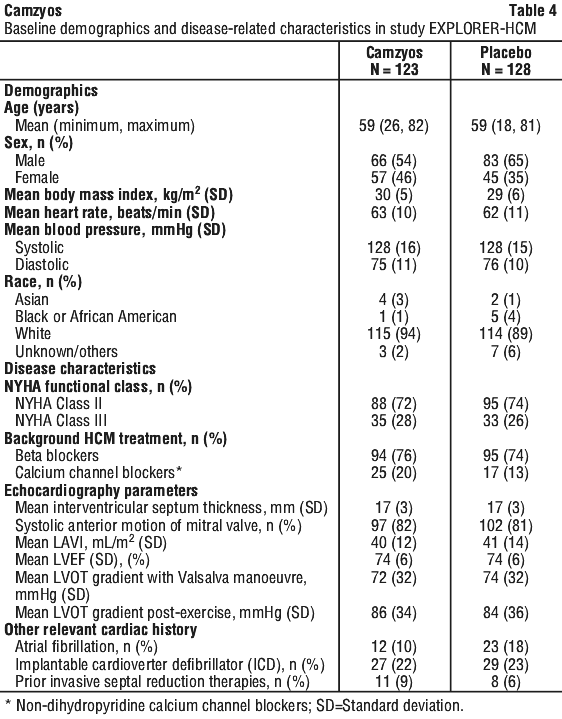
The baseline demographic and disease characteristics are shown in Table 4.
 The primary endpoint was comprised of a composite of change at Week 30 in exercise capacity measured by pVO2 and symptoms measured by NYHA functional classification, defined as an improvement of pVO2 by ≥ 1.5 mL/kg/min and an improvement in NYHA class by at least 1 or an improvement of pVO2 by ≥ 3.0 mL/kg/min and no worsening in NYHA class.
The primary endpoint was comprised of a composite of change at Week 30 in exercise capacity measured by pVO2 and symptoms measured by NYHA functional classification, defined as an improvement of pVO2 by ≥ 1.5 mL/kg/min and an improvement in NYHA class by at least 1 or an improvement of pVO2 by ≥ 3.0 mL/kg/min and no worsening in NYHA class.
A greater proportion of patients met the primary endpoint at Week 30 in the Camzyos arm compared to the placebo arm (36.6% versus 17.2%, respectively, p = 0.0005) (see Table 5).
 A range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications were examined for their influence on outcomes. Results of the primary analysis consistently favoured Camzyos across all subgroups analysed (see Figure 4).
A range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications were examined for their influence on outcomes. Results of the primary analysis consistently favoured Camzyos across all subgroups analysed (see Figure 4).
 The benefit of mavacamten on the primary endpoint was smaller in patients on background beta blocker therapy versus those who were not. Beta blocker therapy blunts heart rate, which attenuates improvements in pVO2. Analyses of other secondary endpoints (symptoms, LVOT gradient) as well as other non-heart rate-dependent cardiopulmonary exercise testing measurements (VE/VCO2 slope) suggest that patients may benefit from mavacamten treatment regardless of beta blocker use.
The benefit of mavacamten on the primary endpoint was smaller in patients on background beta blocker therapy versus those who were not. Beta blocker therapy blunts heart rate, which attenuates improvements in pVO2. Analyses of other secondary endpoints (symptoms, LVOT gradient) as well as other non-heart rate-dependent cardiopulmonary exercise testing measurements (VE/VCO2 slope) suggest that patients may benefit from mavacamten treatment regardless of beta blocker use.
The treatment effects of Camzyos on LVOT obstruction, functional capacity, and health status were assessed by change from baseline through Week 30 in post-exercise LVOT peak gradient, change in pVO2, proportion of patients with improvement in NYHA class, Kansas city cardiomyopathy questionnaire-23 (KCCQ-23) Clinical summary score (CSS), and Hypertrophic cardiomyopathy symptom questionnaire (HCMSQ) Shortness of breath (SoB) domain score. At Week 30, patients receiving Camzyos had greater improvement compared to placebo arm across all secondary endpoints (see Table 6).
 The KCCQ-23 CSS favoured Camzyos compared to placebo at Week 30. The mean improvement from baseline on the KCCQ-23 CSS was greater in the Camzyos arm compared to placebo at Week 30 (p < 0.0001), with effects observed as early as 6 weeks (see Figure 5).
The KCCQ-23 CSS favoured Camzyos compared to placebo at Week 30. The mean improvement from baseline on the KCCQ-23 CSS was greater in the Camzyos arm compared to placebo at Week 30 (p < 0.0001), with effects observed as early as 6 weeks (see Figure 5).
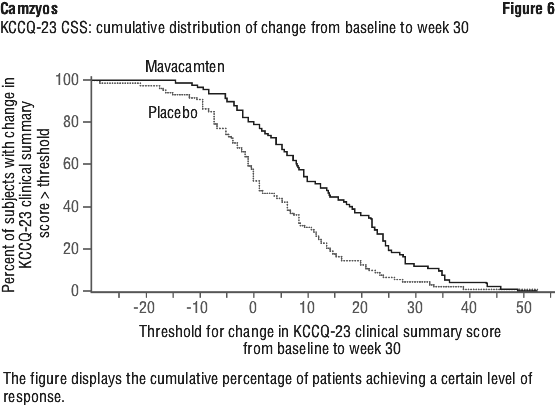
 A greater proportion of patients taking Camzyos compared to placebo achieved a clinically meaningful improvement of ≥ 10 points between baseline and Week 30 in health status (symptoms and physical limitations) (see Figure 6).
A greater proportion of patients taking Camzyos compared to placebo achieved a clinically meaningful improvement of ≥ 10 points between baseline and Week 30 in health status (symptoms and physical limitations) (see Figure 6).
 The HCMSQ SoB domain favoured Camzyos compared to placebo at Week 30. The mean improvement from baseline on the HCMSQ SoB domain was greater in the Camzyos arm compared to placebo at Week 30 (p < 0.0001), with effects observed as early as 4 weeks (see Figure 7).
The HCMSQ SoB domain favoured Camzyos compared to placebo at Week 30. The mean improvement from baseline on the HCMSQ SoB domain was greater in the Camzyos arm compared to placebo at Week 30 (p < 0.0001), with effects observed as early as 4 weeks (see Figure 7).
 A greater proportion of patients taking Camzyos compared to placebo achieved a clinically meaningful improvement of ≤ -2.5 points between baseline and Week 30 in the HCMSQ SoB domain (see Figure 8).
A greater proportion of patients taking Camzyos compared to placebo achieved a clinically meaningful improvement of ≤ -2.5 points between baseline and Week 30 in the HCMSQ SoB domain (see Figure 8).

VALOR-HCM trial.
The efficacy of Camzyos was evaluated in a double-blind, randomized, 16 week placebo-controlled, multicentre, international Phase 3 study (VALOR-HCM) enrolling 112 adult patients with symptomatic obstructive HCM who were eligible for septal reduction therapy (SRT). Patients with severely symptomatic drug-refractory obstructive HCM (including 33% on any combination of beta-blocker, calcium channel blocker and/or disopyramide; 20% on disopyramide alone or in combination with other treatment), and NYHA class II with exertional syncope or near syncope (N=8 [7.1%]), class III (N=103 [92.0%]) or class IV (N=1 [0.9%]), were included in the study. Patients were required to have LVOT peak gradient ≥ 50 mmHg at rest or with provocation, and LVEF ≥ 60%. Patients must have been referred or under active consideration within the past 12 months for SRT and actively considering scheduling the procedure.
Patients were randomized 1:1 to receive either a starting dose of 5 mg of Camzyos (56 patients) or placebo (56 patients) once daily for 16 weeks. Dose adjustment was based on clinical echocardiogram parameters.
Camzyos was shown to be superior to placebo in reducing the proportion of patients who met the primary endpoint (the composite of patient decision to proceed with SRT prior to or at Week 16 or met SRT eligibility (LVOT gradient of ≥ 50 mmHg and NYHA class III-IV, or class II with exertional syncope or near syncope)) at Week 16 (18% vs. 77%, respectively, p < 0.0001; (see Table 7)).
 The treatment effects of Camzyos on LVOT obstruction, functional capacity, health status, and cardiac biomarkers were assessed by change from baseline through Week 16 in post-exercise LVOT gradient, proportion of patients with improvement in NYHA class, KCCQ-23 CSS, NT-proBNP, and cardiac troponin I. In the VALOR-HCM study, hierarchical testing of secondary efficacy endpoints showed significant improvement in the Camzyos group compared to the placebo group (see Table 8).
The treatment effects of Camzyos on LVOT obstruction, functional capacity, health status, and cardiac biomarkers were assessed by change from baseline through Week 16 in post-exercise LVOT gradient, proportion of patients with improvement in NYHA class, KCCQ-23 CSS, NT-proBNP, and cardiac troponin I. In the VALOR-HCM study, hierarchical testing of secondary efficacy endpoints showed significant improvement in the Camzyos group compared to the placebo group (see Table 8).
 The KCCQ-23 CSS favored Camzyos compared to placebo at Week 16. The mean improvement from baseline on the KCCQ-23 CSS was greater in the Camzyos group compared to the placebo at Week 16 (p < 0.0001), with effects observed as early as 4 weeks (see Figure 9).
The KCCQ-23 CSS favored Camzyos compared to placebo at Week 16. The mean improvement from baseline on the KCCQ-23 CSS was greater in the Camzyos group compared to the placebo at Week 16 (p < 0.0001), with effects observed as early as 4 weeks (see Figure 9).
 The proportion of patients with improved KCCQ-23 CSS from baseline to Week 16 was higher at various levels of improvement for the Camzyos treated group compared to the placebo group (see Figure 10 KCCQ-23 Clinical Summary Score: Cumulative Distribution of Change from Baseline to Week 16).
The proportion of patients with improved KCCQ-23 CSS from baseline to Week 16 was higher at various levels of improvement for the Camzyos treated group compared to the placebo group (see Figure 10 KCCQ-23 Clinical Summary Score: Cumulative Distribution of Change from Baseline to Week 16).

5.2 Pharmacokinetic Properties
Mavacamten has a variable terminal t½ that depends on CYP 2C19 metabolic status (6 to 9 days in normal metabolisers and 23 days in poor metabolisers). Exposure to mavacamten increased approximately dose proportionally between 2 mg and 48 mg.
Absorption.
Mavacamten is readily absorbed (Tmax of 1 to 2 hours) after oral administration with an estimated oral bioavailability of approximately 85% within the clinical dose range. The increase in mavacamten exposure is generally dose proportional after once daily doses of mavacamten (2 mg to 48 mg).
Effect of food.
A high fat, high calorie meal delayed absorption resulting in a Tmax of 4 hours in the fed state compared to 1 hour in the fasted state. Administration with food resulted in a 13% decrease in AUC0-inf, however this decrease is not considered clinically significant. Camzyos may be administered with or without food.
Distribution.
Specific studies to assess distribution of mavacamten have not been conducted in humans, however data are consistent with a high volume of distribution. Plasma protein binding of mavacamten is 97-98% in clinical studies. The blood-to-plasma concentration ratio is 0.79.
Based upon measurements of mavacamten in semen of 10 male subjects who received either an 18.5 mg (n=4) or 25 mg (n=6) dose for up to 28 days, the mean (SD) mavacamten semen-to-plasma ratio was 0.039 (0.0047) and 0.044 (0.016), respectively.
Metabolism.
Mavacamten is extensively metabolised, primarily through CYP 2C19 (74%), CYP 3A4 (18%), and CYP 2C9 (7.6%). Three metabolites have been detected in human plasma. The exposure of the most abundant metabolite MYK-1078 in human plasma was less than 4% of the exposure of mavacamten, and the other two metabolites had exposures less than 3% of the exposure of mavacamten indicating these would have minimal to no impact on the overall activity of mavacamten.
Elimination.
Mavacamten is cleared from plasma primarily by metabolism through cytochrome P450 enzymes. Terminal half-life is 6-9 days in CYP 2C19 NM. Drug accumulation occurs with an accumulation ratio about 2-fold for Cmax and about 7-fold for AUC.
At steady-state, the peak-to-trough plasma concentration ratio with once daily dosing is approximately 1.5. Intersubject pharmacokinetic (PK) variability is moderate, with a coefficient of variation for exposure in of approximately 30 to 50% for Cmax and AUC.
CYP 2C19 poor metabolisers.
After a single dose of 15 mg mavacamten, Cmax and AUCinf increased by 47% and 241%, respectively, in CYP 2C19 poor metabolisers (PM) compared to NM. Mean half-life is prolonged in CYP 2C19 PM compared to NM (23 days vs 6 to 9 days, respectively). The incidence of CYP 2C19 PM ranges from approximately 2% in Caucasian to 18% in Asian populations.
Excretion.
Following a single 25 mg dose of 14C labeled mavacamten, 7% and 85% of the total radioactivity was recovered in the faeces and urine, respectively. Unchanged drug accounted for approximately 1% and 3% of the administered dose in the faeces and urine, respectively.
Special populations.
No clinically significant differences in the pharmacokinetics of mavacamten were observed using population PK modelling based on age, sex, race or ethnicity.
Renal impairment.
Approximately 3% of a mavacamten dose is excreted in the urine as parent drug. A population PK analysis, which comprised eGFR levels down to 29.5 mL/min/1.73 m2, demonstrated no correlation between renal function and exposure. A dedicated PK study has not been conducted in patients with severe renal impairment (< 30 mL/min/1.73 m2) (see Section 4.2 Dose and Method of Administration).
Hepatic impairment.
A single dose PK study was conducted in patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment, as well as a control group with normal hepatic function. Mavacamten exposures (AUC) increased 3.2-fold and 1.8-fold in patients with mild and moderate impairment, respectively, compared to patients with normal hepatic function. There was no effect of hepatic function on Cmax, consistent with no change in the rate of absorption and/or volume of distribution. A dedicated PK study has not been conducted in patients with severe (Child-Pugh C) hepatic impairment (see Section 4.2 Dose and Method of Administration).
Elderly.
Clinical trials included 319 patients dosed with Camzyos, 119/319 (37.3%) patients were 65 years of age or older, and 25/319 (7.8%) were age 75 years or older. There were no clinically significant differences observed in safety, efficacy and pharmacokinetics between elderly patients (≥ 65 years) and younger patients (18 to < 65 years) (see Section 4.2 Dose and Method of Administration).
Paediatric and adolescent.
The safety and efficacy of Camzyos in paediatric patients aged less than 18 years of age have not been established. No data are available.
5.3 Preclinical Safety Data
Genotoxicity.
Mavacamten was not found to be genotoxic in a reverse mutation bacterial test (Ames test), a human in vitro lymphocyte clastogenicity assay, or a rat in vivo micronucleus assay.
Carcinogenicity.
There was no evidence of carcinogenicity at the highest mavacamten doses tested in a 6-month rasH2 transgenic mouse study or a 2-year rat study. Exposures (AUC) in mice were up to 3-fold higher compared to the MRHD, while exposures (AUC) in rats were up to 0.2-fold compared to the maximum recommended human dose (MRHD).
Animal toxicology.
The safety of mavacamten has been evaluated in rats and dogs dosed for up to 6 and 9 months, respectively. Noted toxicities, including echocardiographic findings of reduced systolic performance and cardiac dilation, death, due to heart failure, and, in rats, increased heart weights likely secondary to cardiac hypertrophy in response to decreased contractility, were consistent with the mavacamten mechanism of action and primary pharmacological activity. Other findings included cardiac osseous metaplasia in rats and QTc prolongation in dogs. Plasma exposures (AUC) at the no observed adverse effect level NOAEL in rats and dogs respectively are lower than those in humans at the MRHD.6 Pharmaceutical Particulars
6.1 List of Excipients
Capsules.
Silicon dioxide; mannitol; hypromellose; croscarmellose sodium; magnesium stearate.
Capsule shell.
Gelatin; titanium dioxide; black iron oxide; red iron oxide; yellow iron oxide.
Printing ink.
TekPrint SW-9008-N / SW-9009-N black ink.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Polyvinylchloride (PVC)/ polychlorotrifluoroethylene (PCTFE)/ aluminium foil blister containing 14 hard capsules.
Pack size 28 capsules.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
The chemical name of mavacamten is 3-(1-methylethyl)-6- [[(1S)-1-phenylethyl]amino]-2,4(1H,3H)-pyrimidinedione.
The IUPAC name is 6-[[(1S)-1-phenylethyl]amino]- 3-propan-2-yl-1H-pyrimidine-2,4-dione.
The molecular formula is C15H19N3O2, and the molecular weight is 273.33 g/mol.
The chemical structure is:

CAS number.
1642288-47-8.
Mavacamten is a white to off-white powder that is practically insoluble in water and aqueous buffers, sparingly soluble in methanol and ethanol, and freely soluble in dimethyl sulfoxide and N-methyl-2-pyrrolidone.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
