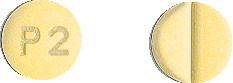

What is in this leaflet
This leaflet answers some common questions about CHOLVASTIN (Pravastatin Sodium).
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking CHOLVASTIN against the benefits it is expected to have for you.
If you have any concerns about using/taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with this medicine. You may need to read it again.
What CHOLVASTIN tablets are used for
CHOLVASTIN tablets belong to a group of lipid-lowering medicines called HMG-CoA reductase inhibitors (commonly known as "statins"). They work by lowering lipids (fats) such as cholesterol and triglycerides in your blood when non-medicinal measures, such as a low-fat diet, exercise and lifestyle changes, alone have failed.
Cholesterol is a naturally occurring substance in the body necessary for normal growth. However, if there is too much cholesterol in your blood, it can get deposited along the walls of the blood vessels leading to narrowing and even blocking of these vessels (atherosclerosis). This is one of the common causes of heart problems such as chest pain (angina) and heart attack (myocardial infarction). High cholesterol levels have been shown to be associated with increased risk of heart disease. Other factors including high blood pressure, high blood sugar (diabetes), increased weight, lack of exercise and smoking further add to the risk of heart disease. High cholesterol levels in the blood can be brought down and kept down with dietary modifications, exercise and medicines.
CHOLVASTIN tablets are used for one or more of the following:
- If you already have heart disease and raised levels of cholesterol, pravastatin will help reduce your chances of having another heart attack, or a stroke. It will reduce your chances of requiring a procedure (such as coronary artery bypass grafting and percutaneous transluminal coronary angioplasty) to increase the blood flow to your heart, and will slow down any further narrowing of your arteries.
- If you do not have heart disease but just raised levels of cholesterol, pravastatin will help prolong your life, reduce the risk of your having a heart attack or requiring a procedure to increase the blood flow to your heart.
- If you have a condition known as primary hypercholesterolaemia, pravastatin will help bring down the cholesterol levels in your blood.
There is no evidence that CHOLVASTIN tablets are addictive.
CHOLVASTIN tablets are not expected to affect your ability to drive a car or operate machinery.
Before you take CHOLVASTIN tablets
When you must not take it
Do not take CHOLVASTIN tablets if you have liver disease or any unexplained abnormal blood test results for liver function.
Do not take CHOLVASTIN tablets if you are allergic to pravastatin or any of the inactive ingredients listed at the end of this leaflet.
Do not take CHOLVASTIN tablets after the expiry date printed on the pack. If you take it after the expiry date has passed, it may not work as well. Do not take CHOLVASTIN if the tablets do not look quite right.
Do not take CHOLVASTIN tablets if the packaging is torn or shows signs of tampering.
Do not give CHOLVASTIN tablets to children under eight years of age.
The safety of CHOLVASTIN tablets in children under 8 years of age has not been studied.
Before you start to take it
Tell your doctor if you have any allergies to:
- any other medicines
- any other substances, such as foods, preservatives or dyes
Tell your doctor if you are pregnant or intend to become pregnant. Like many other medicines, CHOLVASTIN tablets are not recommended during pregnancy. Your doctor will discuss the risks and benefits of using CHOLVASTIN tablets if you are pregnant.
Tell your doctor if you are breast-feeding or planning to breast-feed. A small quantity of pravastatin is known to pass into breast milk. CHOLVASTIN tablets are not recommended while breast-feeding. Your doctor will discuss the possible risks and benefits of taking/using CHOLVASTIN tablets while breast-feeding.
Tell your doctor if you have or have had any medical conditions, especially problems with your kidney(s)/liver.
Tell your doctor if you plan to have surgery.
Tell your doctor if you drink alcohol regularly.
Tell your doctor if you have had muscle pain, tenderness or weakness while taking other medicine used to treat high cholesterol.
If you have not told your doctor about any of the above, tell him/her before you use CHOLVASTIN tablets.
Taking other medicines
Tell your doctor if you are taking any other medicines, including medicines you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines may interfere with the absorption or action of CHOLVASTIN tablets.
These include:
- Gemfibrozil, niacin, colestipol, cholestyramine (other lipid-lowering medicines)
- Cyclosporin (a medicine which modulates immune response)
- Erythromycin (antibacterial medicine)
- Ketoconazole (antifungal medicine)
- Cimetidine (medicine used for healing peptic ulcers)
- Spironolactone (potassium-sparing water tablets)
These medicines may be affected by CHOLVASTIN, or may affect how well it works. You may need to use different amounts of your medicine or you may need to take different medicines. Your doctor or pharmacist will be able to tell you what to do when taking/being given CHOLVASTIN tablets with other medicines.
Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking CHOLVASTIN tablets.
How CHOLVASTIN tablets are given
How much to take
Make sure you continue with your recommended diet, exercise and other lifestyle changes while on your medicine.
How to take it
Swallow the tablet whole with a full glass of water.
For Hypercholesterolaemia
Adults
The usual starting dose is one half or one CHOLVASTIN 20 mg tablet taken once daily, at bedtime. Your doctor may adjust the dosage, depending on your response to the medicine.
The recommended dose of pravastatin could vary from 10 mg to 80 mg per day, taken once daily at bedtime or taken evenly in divided doses.
Adolescents (14 to 18 years)
The usual recommended dose is pravastatin 40 mg taken once daily.
Children (8 to 13 years)
The usual recommended dose is pravastatin 20 mg taken once daily.
The action of CHOLVASTIN tablets starts gradually and it may take up to four weeks to establish control of your blood cholesterol levels. Once your blood cholesterol is controlled, your doctor may reduce your daily dose to suit your condition.
For prevention of heart disease or stroke
The usual dose is one CHOLVASTIN 40 mg tablet taken once daily.
Your doctor may initially prescribe lower doses for you if you are elderly, or if you have kidney or liver problems, or you are taking other medicines such as immune system modulators if other lipid- lowering agents.
When to take it
For achieving maximum benefits from CHOLVASTIN tablets, take them preferably at bedtime on an empty stomach.
Take your CHOLVASTIN tablets at about the same time each day. Taking your tablets at the same time each day will have the best effect. It will also help you to remember when to take the tablets.
How long to take it
Continue taking CHOLVASTIN tablets until your doctor tells you to stop. Do not stop taking CHOLVASTIN tablets suddenly. CHOLVASTIN tablets help control high blood cholesterol levels as well as the associated long-term complications. Therefore you must take CHOLVASTIN tablets every day and for a period as long as advised by your doctor. Most people will need to take CHOLVASTIN tablets for long periods of time.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, then go back to taking it as you would normally.
Do not double a dose to make up for the dose you have missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much (Overdose)
Immediately telephone your doctor or the Poisons Information Centre (13 11 26), or go to Accident and Emergency at your nearest hospital, if you think that you or anyone else has taken too much of CHOLVASTIN tablets. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention. Keep this telephone number handy.
While you are using CHOLVASTIN tablets
Things you must remember
Take CHOLVASTIN tablets exactly as your doctor tells you to.
Try not to miss any dose and take the medicine even if you feel well.
Visit your doctor regularly for check-ups and monitor your blood cholesterol levels as instructed by your doctor.
You should monitor your blood cholesterol levels regularly to make sure that CHOLVASTIN tablets are controlling your cholesterol levels.
Tell all doctors, dentists and pharmacists who are treating you that you are taking CHOLVASTIN tablets.
If you are about to be started on any new medicine, tell your doctor or pharmacist that you are taking CHOLVASTIN tablets.
If you plan to have surgery make sure you tell your doctor, dentist or anaesthetist that you are taking CHOLVASTIN tablets.
If you become pregnant while taking CHOLVASTIN tablets, tell your doctor immediately.
Things you must not do
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not use CHOLVASTIN tablets to treat any other complaints unless your doctor tells you to.
Do not stop taking CHOLVASTIN tablets, or lower the dose, without first checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how CHOLVASTIN tablets affect you. CHOLVASTIN tablets are not known to affect your ability to drive or operate machinery. However, they may make you feel dizzy which could affect your ability to drive or operate machinery. Make sure you know how you react to CHOLVASTIN tablets before you drive a car, operate machinery, or do anything else that could be dangerous if you are not alert.
Thing that may help your condition
- Diet
Eat a healthy diet, which includes plenty of fresh vegetables, fruit, bread, cereals and fish. Also eat less fat and sugar. - Exercise
Regular exercise helps reduce blood pressure and helps the heart get fitter, but it is important not to overdo it. Walking is good exercise, but try to find a route that is fairly flat. Before starting any exercise, ask your doctor about the best kind of programme for you. - Smoking
Your doctor may advise you to stop or at least cut down smoking. - Weight
Your doctor may suggest losing weight to help lower your blood pressure and help lessen the amount of work your heart has to do. Some patients may need to see a dietician to help lose weight.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking CHOLVASTIN tablets.
Pravastatin helps most people to reduce increased blood cholesterol levels, but may have unwanted side effects in a few people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- Feeling sick (nausea) or being sick (vomiting), diarrhoea or loose stools, indigestion or heartburn, escape of wind, constipation, abdominal pain and bloating
- Tiredness, weakness, chest pain, weight gain/loss
- Muscle pain or cramps
- Dizziness, headache, sleep disturbance, depressed mood, anxiety/nervousness, numbness, tingling or burning sensation or pain in the feet and/or hands, uncontrolled shaking
- Loss of hair, any other changes in skin/nail/hair, increased sensitivity to sunlight
- Altered taste sensation, worsening of pre-existing cloudiness in the lens of the eye
- Persistent abnormal erection of the penis, decreased sexual performance including impaired ejaculation in males and decreased orgasm
- Breast enlargement in male or abnormal production of breast milk
- Cough, running nose
These are the more common side effects of CHOLVASTIN tablets. These side effects are usually mild and occur at the start of treatment.
Tell your doctor immediately if you notice any of the following:
- Unexplained/persistent muscle aches, with or without passage of pink coloured urine
- Unusual bleeding or increased tendency to bleed, persistent sore throat and frequent infections, and/or anaemia
- Sudden onset of severe abdominal pain associated with feeling sick (nausea) and being sick (vomiting)
- Yellowing of skin and whites of eyes with decreased appetite, abdominal pain
- Recurrent episodes of numbness and loss of muscle strength (pressure palsies)
- Buzzing or ringing in the ears
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- Symptoms of an allergic reaction may include rashes, hives, itching, chest constriction, shortness of breath or swelling of face, lips, tongue, hands/feet, fainting, fever.
These are very serious side effects. You may need urgent medical attention or hospitalisation. All these side effects are very rare.
Statins may very rarely be associated with a type of lung disease. You should see your doctor if you experience breathlessness or difficulty breathing, a cough and deterioration in your general health (e.g. weight loss, fever, fatigue) while taking this medicine.
Other side effects not listed above may also occur in some patients. Tell you doctor if you notice anything else that is making you feel unwell. Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using it
Storage
Keep your tablets in the blister pack until it is time to take them. If you take the tablets out of the box or the blister pack they may not keep well.
Keep your CHOLVASTIN tablets in a cool, dry place where it stays below 25°C.
Do not store it in the bathroom or near a sink.
Do not leave it in the car on hot days. Heat or exposure to light and dampness can destroy medicines.
Keep this medicine where young children cannot reach it.
Disposal
If your doctor tells you to stop taking or you find that the tablets have passed their expiry date, ask your pharmacist what to do with any tablets that are left over.
Product description
What it looks like
- CHOLVASTIN 20 mg are dark yellow to yellow coloured mottled, circular, biconvex uncoated tablets debossed with ‘P2’ on one side and a breakline on the other side. Available in packs of 30 tablets.
- CHOLVASTIN 40 mg are dark yellow to yellow coloured mottled, circular, biconvex uncoated tablets debossed with ‘P3’ on one side and a breakline on the other side. Available in packs of 30 tablets.
Ingredients
Active ingredient
- CHOLVASTIN 20 mg tablets: 20 mg pravastatin sodium
- CHOLVASTIN 40 mg tablets: 40 mg pravastatin sodium
Inactive ingredients
- Lactose anhydrous
- Sodium stearyl fumarate
- Ferric oxide yellow
Sponsor
Ranbaxy Australia Pty Ltd.
Suite 4.02, Building D, Level 4
12 – 24 Talavera Road
North Ryde NSW 2113
Australia
CHOLVASTIN is supplied in New Zealand by:
Douglas Pharmaceuticals Ltd
Central Park Drive, Lincoln
Auckland 0610
New Zealand
Australian Registration Numbers
- CHOLVASTIN 20 mg Tablets: AUST R 140038
- CHOLVASTIN 40 mg Tablets: AUST R 140042
This leaflet was prepared in May 2011.
Published by MIMS December 2011

 In a pooled analysis of two multicenter, double blind, placebo controlled studies in patients with primary hypercholesterolemia, treatment with pravastatin at a daily dose of 80 mg increased HDL-C and significantly decreased total-C, LDL-C, and TG from baseline after 6 weeks. The efficacy results of the individual studies were consistent with the pooled data. Mean percent changes from baseline after 6 weeks of treatment were: total-C (-27%), LDL-C (-37%), HDL-C (+3%) and TG (-19%), with placebo subtracted changes for LDL-C and TG of -36% and -20%, respectively.
In a pooled analysis of two multicenter, double blind, placebo controlled studies in patients with primary hypercholesterolemia, treatment with pravastatin at a daily dose of 80 mg increased HDL-C and significantly decreased total-C, LDL-C, and TG from baseline after 6 weeks. The efficacy results of the individual studies were consistent with the pooled data. Mean percent changes from baseline after 6 weeks of treatment were: total-C (-27%), LDL-C (-37%), HDL-C (+3%) and TG (-19%), with placebo subtracted changes for LDL-C and TG of -36% and -20%, respectively. The effect on the combined endpoint of coronary heart disease death or nonfatal myocardial infarction was evident as early as six months after beginning pravastatin therapy.
The effect on the combined endpoint of coronary heart disease death or nonfatal myocardial infarction was evident as early as six months after beginning pravastatin therapy.
 In the Long-Term Intervention with Pravastatin in Ischemic Disease (LIPID) study, the effect of pravastatin 40 mg daily was assessed in 9014 men and women with normal to elevated serum cholesterol levels (baseline total-C = 4.0-7.0 mmol/L; mean total-C = 5.66 mmol/L; mean total-C/HDL-C ratio = 5.9), and who had experienced either a myocardial infarction or had been hospitalised for unstable angina pectoris in the preceding 3-36 months. Patients with a wide range of baseline levels of triglycerides were included (≤ 5.0 mmol/L) and enrollment was not restricted by baseline levels of HDL cholesterol. At baseline, 82% of patients were receiving aspirin, 76% were receiving antihypertensive medication, and 41% had undergone myocardial revascularisation. Patients in this multicentre, double blind, placebo controlled study participated for a mean of 5.6 years (median = 5.9 years). Treatment with pravastatin significantly reduced the risk for CHD death by 24% (p = 0.0004). The risk for coronary events (either CHD death or nonfatal MI) was significantly reduced by 24% (p < 0.0001) in the pravastatin treated patients. The risk for fatal or nonfatal myocardial infarction was reduced by 29% (p < 0.0001). Pravastatin reduced both the risk for total mortality by 23% (p < 0.0001) and cardiovascular mortality by 25% (p < 0.0001). The risk for undergoing myocardial revascularisation procedures (coronary artery bypass grafting or percutaneous transluminal coronary angioplasty) was significantly reduced by 20% (p < 0.0001) in the pravastatin treated patients. Pravastatin also significantly reduced the risk for stroke by 19% (p = 0.0477). Treatment with pravastatin significantly reduced the number of days of hospitalisation per 100 person years of follow-up by 15% (p < 0.001). The prespecified subgroup (age, sex, hypertensives, diabetics, smokers, lipid subgroups) analyses were conducted using the combined end point of CHD and nonfatal MI. The study was not powered to examine results within each subgroup but formal testing for heterogeneity of treatment effect was undertaken across each of the subgroups and no significant heterogeneity was found (p ≥ 0.08), i.e. a consistent treatment effect was seen with pravastatin therapy across all patient subgroups and event parameters. Among patients who qualified with a history of myocardial infarction, pravastatin significantly reduced the risk for total mortality by 25% (p = 0.0016); for CHD mortality by 23% (p = 0.004); for CHD events by 22% (p = 0.002) and for fatal or nonfatal MI by 25% (p = 0.0008). Among patients who qualified with a history of hospitalisation for unstable angina pectoris, pravastatin significantly reduced the risk for total mortality by 26% (p = 0.0035; for CHD mortality by 26% (p = 0.0358); for CHD events by 29% (p = 0.0001) and for fatal or nonfatal MI by 37% (p = 0.0003). (See Table 5.)
In the Long-Term Intervention with Pravastatin in Ischemic Disease (LIPID) study, the effect of pravastatin 40 mg daily was assessed in 9014 men and women with normal to elevated serum cholesterol levels (baseline total-C = 4.0-7.0 mmol/L; mean total-C = 5.66 mmol/L; mean total-C/HDL-C ratio = 5.9), and who had experienced either a myocardial infarction or had been hospitalised for unstable angina pectoris in the preceding 3-36 months. Patients with a wide range of baseline levels of triglycerides were included (≤ 5.0 mmol/L) and enrollment was not restricted by baseline levels of HDL cholesterol. At baseline, 82% of patients were receiving aspirin, 76% were receiving antihypertensive medication, and 41% had undergone myocardial revascularisation. Patients in this multicentre, double blind, placebo controlled study participated for a mean of 5.6 years (median = 5.9 years). Treatment with pravastatin significantly reduced the risk for CHD death by 24% (p = 0.0004). The risk for coronary events (either CHD death or nonfatal MI) was significantly reduced by 24% (p < 0.0001) in the pravastatin treated patients. The risk for fatal or nonfatal myocardial infarction was reduced by 29% (p < 0.0001). Pravastatin reduced both the risk for total mortality by 23% (p < 0.0001) and cardiovascular mortality by 25% (p < 0.0001). The risk for undergoing myocardial revascularisation procedures (coronary artery bypass grafting or percutaneous transluminal coronary angioplasty) was significantly reduced by 20% (p < 0.0001) in the pravastatin treated patients. Pravastatin also significantly reduced the risk for stroke by 19% (p = 0.0477). Treatment with pravastatin significantly reduced the number of days of hospitalisation per 100 person years of follow-up by 15% (p < 0.001). The prespecified subgroup (age, sex, hypertensives, diabetics, smokers, lipid subgroups) analyses were conducted using the combined end point of CHD and nonfatal MI. The study was not powered to examine results within each subgroup but formal testing for heterogeneity of treatment effect was undertaken across each of the subgroups and no significant heterogeneity was found (p ≥ 0.08), i.e. a consistent treatment effect was seen with pravastatin therapy across all patient subgroups and event parameters. Among patients who qualified with a history of myocardial infarction, pravastatin significantly reduced the risk for total mortality by 25% (p = 0.0016); for CHD mortality by 23% (p = 0.004); for CHD events by 22% (p = 0.002) and for fatal or nonfatal MI by 25% (p = 0.0008). Among patients who qualified with a history of hospitalisation for unstable angina pectoris, pravastatin significantly reduced the risk for total mortality by 26% (p = 0.0035; for CHD mortality by 26% (p = 0.0358); for CHD events by 29% (p = 0.0001) and for fatal or nonfatal MI by 37% (p = 0.0003). (See Table 5.)
 The safety and efficacy of pravastatin doses above 40 mg daily have not been studied in children. The long-term efficacy of pravastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
The safety and efficacy of pravastatin doses above 40 mg daily have not been studied in children. The long-term efficacy of pravastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
 There was no statistically significant difference in the change in lens opacity between the control and pravastatin treatment groups during this time interval.
There was no statistically significant difference in the change in lens opacity between the control and pravastatin treatment groups during this time interval.