What is in this leaflet
This leaflet answers some common questions about CIMZIA.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking CIMZIA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What CIMZIA is used for
CIMZIA is used for the treatment of the following:
Rheumatoid arthritis
Rheumatoid arthritis is an inflammatory disease of the joints. Symptoms include joint pain, tenderness, swelling and stiffness.
Psoriatic arthritis
Psoriatic arthritis is an inflammatory disease of the joints and skin, with similarities to rheumatoid arthritis as well as psoriasis.
Ankylosing Spondylitis and Non-radiographic Axial Spondyloarthritis
Ankylosing Spondylitis and Non-radiographic Axial Spondyloarthritis are inflammatory diseases of the spine. Symptoms include back pain and morning stiffness.
Plaque psoriasis
Plaque psoriasis is an inflammatory disease of the skin. Symptoms include red, itchy and inflamed skin.
The active ingredient in CIMZIA, certolizumab pegol, is a humanised monoclonal antibody produced by cultured cells. Monoclonal antibodies are proteins that recognise and bind to other unique proteins.
Over production of the naturally occurring protein, tumour necrosis factor or TNFα is thought to cause rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis and plaque psoriasis. Certolizumab pegol works by binding to a specific protein, TNFα and blocking its action. CIMZIA can prevent the harmful effects of TNFα, thereby reducing the signs and symptoms of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis and plaque psoriasis which include joint pain, tenderness, swelling and stiffness.
CIMZIA is normally used together with another medicine called methotrexate to treat your rheumatoid arthritis.
CIMZIA may be used without methotrexate if your doctor determines that it is unsuitable for you to use or continue methotrexate.
Your doctor may prescribe CIMZIA in addition to your current therapy.
Ask your doctor if you have any questions about why CIMZIA has been prescribed for you.
There is no evidence that CIMZIA is addictive.
This medicine is available only with a doctor’s prescription.
The safety and effectiveness of CIMZIA has not been established in children.
Before you use CIMZIA
When you must not use it
Do not use CIMZIA if you have:
- an allergy to mouse proteins or any of the ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction to CIMZIA may include:
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
- Tuberculosis, other infection such as blood infection, a chronic infection or a history of recurrent infection.
Symptoms of infection may include:
- fever
- wounds
- feeling tired
- dental problems
- Moderate to severe heart failure.
- Do not use CIMZIA if you are already using anakinra or abatacept - medicines for rheumatoid arthritis.
- Do not use CIMZIA after the expiry date (EXP) printed on the pack.
- Do not use CIMZIA if the packaging is torn or shows signs of tampering or if the pre-filled syringe or pen does not look quite right.
If CIMZIA has expired or is damaged, return to your pharmacist for disposal.
Before you use it
Tell your doctor or pharmacist if you have allergies to:
- any other medicines or any other substances, such as foods, preservatives or dyes.
Tell your doctor if you have or have had any medical conditions, especially the following:
- any symptoms of an infection including fever, cough, or any flu-like symptoms
- any open cuts or sores on your body
- have or have had a hepatitis B infection. CIMZIA may increase the risk of reactivation in people who carry this virus
- a history of recurrent infections or other conditions that increase the risk of infections
- nervous system disorder such as multiple sclerosis and other demyelinating disease
- congestive heart failure
- cancer (such as lymphoma and skin cancer)
- symptoms of lupus (persistent rash, fever, joint pain, and tiredness)
- experience allergic reactions such as chest tightness, wheezing, dizziness, swelling or rash
- airway disease such as emphysema or chronic obstructive pulmonary disease
- blood disorder or symptoms of a blood disorder such as persistent bruising, bleeding, paleness or fever
- skin condition such as psoriasis
- tuberculosis, or if you have been in close contact with someone who has had tuberculosis.
As cases of tuberculosis have been reported in patients treated with CIMZIA, your doctor will check you for signs and symptoms of tuberculosis before starting CIMZIA. This will include a thorough medical history, a chest X-ray and a tuberculin test.
Tell your doctor if you are scheduled for any vaccines. Some vaccines should not be given while receiving CIMZIA. Check with your doctor before you receive any vaccines.
Tell your doctor if you are scheduled for any surgery. Talk to your doctor if you are going to have any operations or dental procedures.
Tell your doctor if you are pregnant or plan to become pregnant. Discuss with your doctor on the use of CIMZIA and contraception in pregnancy.
Tell your doctor if you are breastfeeding or plan to breastfeed. Cimzia can be used during breastfeeding.
If you have not told your doctor or pharmacist about any of the above, tell them before you start using CIMZIA.
If you are not sure whether you should start using CIMZIA, talk to your doctor or pharmacist.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop. Some medicines and CIMZIA may interfere with each other. Your doctor and pharmacist have more information on medicines to be careful with or avoid while using this medicine.
Tell your doctor or pharmacist if you are taking other biologic medicines such as anakinra or abatacept. Taking other biologic medicines while on anti-TNF therapy may increase the risk of infection.
CIMZIA can be taken together with methotrexate, antibiotics, corticosteroids, such as prednisone or pain medications including non-steroidal anti-inflammatory drugs.
How to use CIMZIA
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the label or in this leaflet, ask your doctor or pharmacist for help.
How much to use
Always use CIMZIA exactly as your doctor has instructed you. You should check with your doctor or pharmacist if you are unsure.
Rheumatoid Arthritis, Psoriatic arthritis, Ankylosing spondylitis and Non-radiographic Axial Spondyloarthritis
The starting dose is 400 mg CIMZIA (as 2 injections in one day) given at weeks 0, 2 and 4.
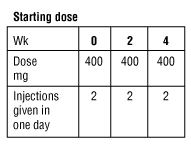
Maintenance dose
This is followed by a maintenance dose of 200 mg every other week starting at week 6. Alternatively, your doctor may decide that you should receive a monthly dose of 400 mg CIMZIA (as 2 injections in one day).
Plaque Psoriasis
The starting dose is 400 mg CIMZIA (as 2 injections in one day) given at weeks 0, 2 and 4, then followed by a maintenance dose of either 200 mg every other week or 400 mg every other week starting at week 6.
Methotrexate is continued while using CIMZIA. If your doctor determines that methotrexate is inappropriate, CIMZIA can be given alone.
Your doctor will tell you how much CIMZIA you will need to use.
If you do not understand the instructions on the pack, ask your doctor or pharmacist for help.
How to use it
CIMZIA is injected under the skin (subcutaneous use). The injection can be self-administered or given by another person, for example a family member or friend after proper training in injection technique, by your doctor or a nurse.
Instructions for preparing and giving an injection of CIMZIA
The following instructions explain how to inject CIMZIA. Read the instructions carefully and follow them step by step.
Do not attempt to self-inject until you are sure that you understand how to prepare and give the injection. Your doctor or a nurse will show you how to prepare and self-inject.
Do not mix the injection in the same syringe or pen with any other medicine.
- Setting up
- Take the carton containing CIMZIA out of the refrigerator.
- Wash your hands thoroughly.
- Set up the following items on a clean surface.
- One pre-filled syringe or pen of CIMZIA
- One alcohol pad - Look at the expiry date on the syringe/pen. Do not use the product after the month and year shown.
- Allow CIMZIA to reach room temperature. This will take about 30 minutes for the pre-filled syringe and 30-45 minutes for the pre-filled pen minutes.
- Do not try to warm up the syringe/pen.
- Choosing and preparing an injection site.
- Choose a site on your thigh or stomach.
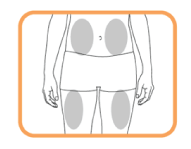
Each new injection should be given on a separate site from the last injection site.
- Do not inject in an area where the skin is tender, reddened, bruised, or hard.
- Wipe the injection site with the enclosed alcohol pad, using a circular motion.
- Do not touch the site again before injecting.
- Injecting CIMZIA
Pre-filled syringe
- Do NOT shake the syringe.
- Check the medicine in the barrel of the syringe is clear to pale yellow and free from particles. Do not use the pre-filled syringe if the solution is discoloured, cloudy or if you can see particles in it. You may see an air bubble. This is normal. There is no need to remove air bubbles before injection. Injecting the solution subcutaneously with air bubbles is harmless.
- Remove the cap from needle syringe by pulling off the plastic ring in a straight line, being careful not to touch the needle or let the needle touch any surface. Do not bend the needle.
- Hold the syringe with the needle facing down.
- With one hand, gently grasp the cleaned areas of skin and hold firmly.
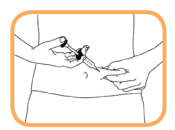
- With the other hand, hold syringe at a 45 degree angle to skin.
- With one quick, short motion, push the needle all the way into the skin.
- Push the plunger to inject solution- it can take up to 10 seconds to empty the syringe.
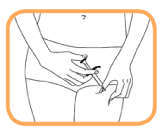
- When the syringe is empty, carefully remove the needle from the skin at the same angle at which it was inserted.
- Release the skin with the first hand.
- Using your thumb or a piece of gauze, apply pressure over the injection site for a few seconds. Do not rub the injection site. You may cover the injection site with a small adhesive bandage, if necessary.
Pre-filled pen
- The pre-filled pen is designed to work accurately and safely. However, if any of the following steps go wrong and/or if you feel unsure about the injection process, contact your doctor or pharmacist.
- Do NOT shake the pre-filled pen.
- Check the medicine in the pre-filled pen (through the viewing window) is clear to pale yellow and free from particles. Do not use the pre-filled pen if the solution is discoloured, cloudy or if you can see particles in it. You may see air bubbles. This is normal. There is no need to remove air bubbles before injection. Injecting the solution subcutaneously with air bubbles is harmless.
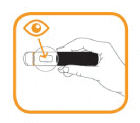
- Hold the pre-filled pen firmly with one hand around the black handle.
- Grasp the clear cap with the other hand and remove it. Do not twist the cap off as this may jam the internal mechanism.
- Inject within 5 minutes of removing the cap. Do not replace the cap.
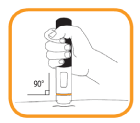
- Although hidden from view the needle tip is now uncovered. Do not try to touch the needle as it could activate the pre-filled pen.
- Hold the pre-filled pen straight (at a 90 degree angle) against the skin that previously has been cleaned (the “injection site”).
- Press the pre-filled pen firmly against the skin and hold. The injection begins when a first “click” is heard and the orange band at the bottom of the pre-filled pen disappears.
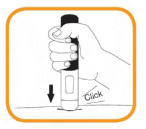
- Continue to hold the pre-filled pen in place firmly against the skin until a second “click” is heard and the viewing window turns orange. This can take up to 15 seconds. At this time, the injection will be complete. If the viewing window turns orange and you hear a second click this means the injection has been completed. If you feel unsure about the injection process, please contact your doctor or pharmacist. Do not try to repeat the injection without speaking to your doctor or your pharmacist.
- The needle will automatically move back into the empty pen. Do not try to touch the needle.
- You can now remove the used pen by pulling the pen straight up carefully from the skin.
- Use a piece of gauze, apply pressure over the injection site for a few seconds. Do not rub the injection site. You may cover the injection site with a small adhesive bandage, if necessary.
- Throwing away supplies
- You must NOT re-use or re-cap the CIMZIA syringe/pen. This product is for one dose in one patient only.
- After injecting CIMZIA, immediately throw away the used syringe/pen in a special container as instructed by your doctor, nurse or pharmacist.
- Keep this container out of the reach of children.
If you forget to use it
If you forget to give yourself an injection, you should inject the next dose of CIMZIA as soon as you remember. Then inject your next dose as you would have on your originally scheduled day, had you not forgotten a dose.
Do not use a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to use your medicine, ask your pharmacist for some hints.
How long to use it
Continue taking CIMZIA for as long as your doctor tells you to. CIMZIA helps control your condition, but does not cure it.
Do not stop taking CIMZIA without talking to your doctor first.
If you use too much (overdose)
If you accidentally inject CIMZIA more frequently than told to by the doctor, immediately telephone your doctor or the Poisons Information Centre, Telephone 13 11 26, or go to Accident and Emergency at your nearest hospital. Do this even if there are no signs of discomfort or poisoning. Always take the outer carton of the medicine with you.
While you are using CIMZIA
Things you must do
Check with your doctor before you receive any vaccines.
Tell any other doctors, dentists, and pharmacists who are treating you that you are using CIMZIA.
If you are about to be started on any new medicine, tell your doctor, dentist or pharmacist that you are using CIMZIA.
Before you have any surgery or emergency treatment, tell your doctor or dentist that you are using CIMZIA.
Tell your doctor if you feel CIMZIA is not helping your condition. Your doctor may need to change your medicine.
Tell your doctor if, for any reason, you have not used CIMZIA exactly as prescribed. Otherwise, your doctor may change your treatment unnecessarily.
If you become pregnant while taking CIMZIA, tell your doctor.
Be sure to keep all of your doctor’s appointments so that your progress can be checked. Your doctor will check your progress and may want to do some tests from time to time. This helps to prevent unwanted side effects.
Things you must not do
Do not give CIMZIA to anyone else, even if their symptoms seem similar to yours or they have the same condition as you.
Do not use CIMZIA to treat any other complaints unless your doctor tells you to.
Do not use CIMZIA and anakinra together.
Do not use CIMZIA and abatacept together.
Do not stop using it unless your doctor tells you to.
Things to be careful of
Be careful driving or operating machinery until you know how CIMZIA affects you.
The effects on your ability to drive and use machines whilst using CIMZIA are not known.
Make sure you know how you react to CIMZIA before you drive a car, operate machinery, or do anything else that could be dangerous if you are drowsy.
Side effects
Check with your doctor as soon as possible if you have any problem while using CIMZIA, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
Ask your doctor or pharmacist any questions you may have. Like all medicines CIMZIA can cause some side effects, although not everybody gets them.
Tell your doctor immediately if you notice any of the following or go to Accident and Emergency at your nearest hospital:
- severe rash hives or other signs of allergic reaction
- swollen face, hands, feet
- trouble breathing, swallowing
- shortness of breath with exertion or upon lying down or swelling of the feet
- persistent fever, bruising, bleeding, paleness.
These are very serious side effects. You may need urgent medical attention or hospitalisation. These side effects are rare.
Tell your doctor as soon as possible if you notice any of the following:
- feeling weak or tired
- persistent cough, weight loss, listlessness, fever
- signs of nervous system disorders such as numbness or tingling throughout your body, arm or leg weakness, double vision
- a bump or open sore that doesn't heal.
These are serious side effects. You may need urgent medical attention.
Tell your doctor or pharmacist if you notice any of the following:
- lower respiratory tract infections (such as bronchitis, pneumonia)
- upper respiratory infections (such as cold, runny nose, sinus infections)
- bacterial infections in any site, including abscess (a collection of pus) or a urinary tract infection
- viral infections (including cold sores, shingles, and influenza)
- fever
- high blood pressure
- rash or itching
- abdominal symptoms such as abdominal pain, nausea, vomiting, diarrhoea, constipation, pain, distension, heartburn, or indigestion
- injection site reactions (including pain, swelling, redness or itching)
- feeling weak and generally unwell
- headache dizziness.
These are the more common side effects of CIMZIA.
Other side effects not listed above may happen in some people.
There have been cases of certain kinds of cancer in patients using CIMZIA or other TNF blockers. People with more serious rheumatoid arthritis that have had the disease for a long time may have a higher chance of getting a kind of cancer that affects the lymph system, called lymphoma. Talk to your doctor if you are concerned about this.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Tell your doctor if you notice anything else that is making you feel unwell. Some of these side effects can only be found when your doctor does tests from time to time to check your progress.
After using CIMZIA
Storage
Keep your pre-filled syringe or pen in the pack until it is time to use it.
Keep CIMZIA in a refrigerator between 2°C and 8°C. Do not freeze. Protect from light.
If needed, CIMZIA may be stored at room temperature up to a maximum of 25°C for a single period of up to 10 days with protection from light. Once CIMZIA has been stored at room temperature, it should not be placed back into the refrigerator and should be discarded if not used within the 10-day period.
Keep CIMZIA in a place in the refrigerator where children cannot reach it.
Disposal
After injecting CIMZIA, immediately throw away the used syringe or pen in a special container as instructed by your doctor, nurse or pharmacist.
If your doctor or pharmacist tells you to stop using CIMZIA or the medicine has passed its expiry date, ask your pharmacist what to do with any that is left over.
Product description
What it looks like
CIMZIA is a clear to opalescent, colourless to yellow, essentially free of visible particles, sterile solution in a syringe or pen.
CIMZIA is supplied in a carton containing:
- Two 1 mL pre-filled syringes or pre-filled pens of CIMZIA
- Two alcohol wipes (for cleansing the areas chosen for injection)
Ingredients
Each 1mL syringe contains 200 mg certolizumab pegol.
Other ingredients in CIMZIA sterile solution include:
- Sodium acetate
- Sodium chloride
- Water for injection
The needle shield for the Pre-filled syringe and the Pre-filled pen contains epoxyprene, a derivative of natural rubber latex. The needle shield does not come into direct contact with you or the person giving the injection.
Sponsor:
UCB Pharma
A division of UCB Australia Pty Ltd
Level 1, 1155 Malvern Road
Malvern VIC 3144, Australia
CIMZIA 200mg/mL Solution for Injection – pre-filled syringe – AUST R 154726
CIMZIA 200mg/mL Solution for Injection – pre-filled pen – AUST R 281317
Date of preparation:
March 2020
Everyday Support Program
The Everyday Support Program provides a comprehensive resource to assist patients prescribed CIMZIA and offers:
- home visits from a qualified nurse to provide self-injection education
- educational resources.
Call 1800-CIMZIA
(1800 246 942)
Published by MIMS June 2020
 However, patients receiving Cimzia and concomitant methotrexate had a lower humoral response compared with patients receiving Cimzia alone (see Table 2). The clinical significance of this is unknown.
However, patients receiving Cimzia and concomitant methotrexate had a lower humoral response compared with patients receiving Cimzia alone (see Table 2). The clinical significance of this is unknown. Cimzia does not significantly suppress the protective humoral immune response to the pneumococcal polysaccharide vaccine or influenza vaccine.
Cimzia does not significantly suppress the protective humoral immune response to the pneumococcal polysaccharide vaccine or influenza vaccine.
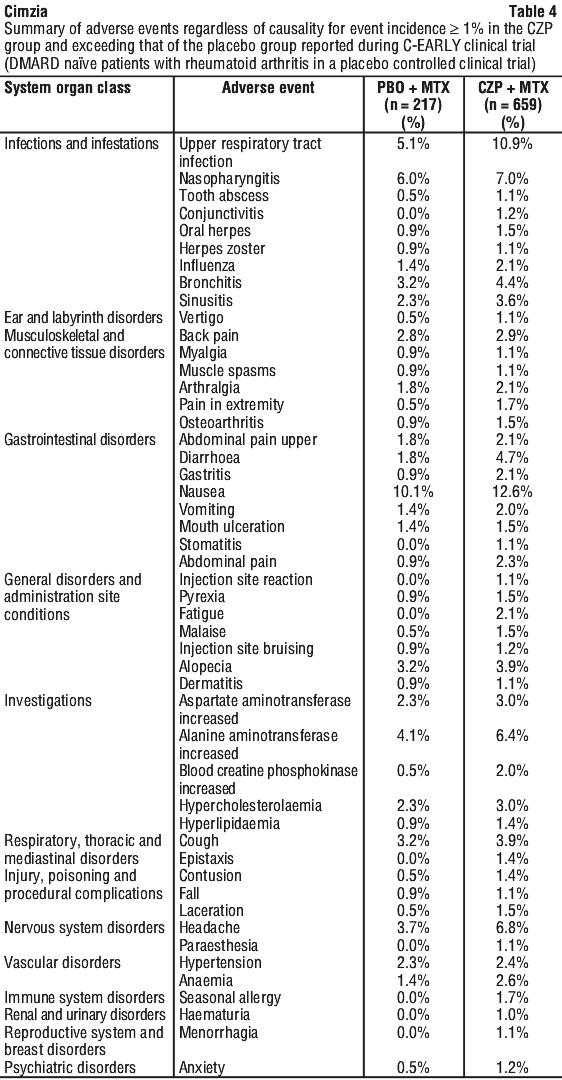

 Within the organ system classes, adverse reactions by frequency are listed using the following categories: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1000), not known (cannot be estimated from the available data) in Table 7.
Within the organ system classes, adverse reactions by frequency are listed using the following categories: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1000), not known (cannot be estimated from the available data) in Table 7. The additional following Adverse Drug Reactions (ADRs) have been observed uncommonly with Cimzia in other indications: gastrointestinal stenosis and obstructions, general physical health deterioration, grand mal convulsion, optic neuritis, abortion spontaneous and azoospermia, vaginal discharge and fistula (any site).
The additional following Adverse Drug Reactions (ADRs) have been observed uncommonly with Cimzia in other indications: gastrointestinal stenosis and obstructions, general physical health deterioration, grand mal convulsion, optic neuritis, abortion spontaneous and azoospermia, vaginal discharge and fistula (any site).
 See Table 10.
See Table 10.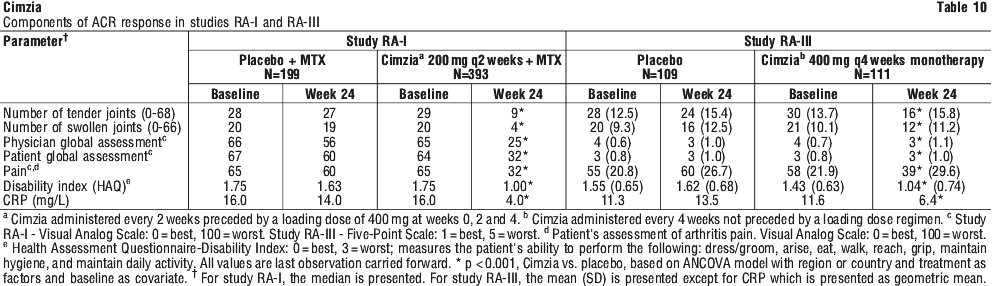 The percentage of patients achieving ACR20 response by visit for Study RA-I is shown in Figure 1. Among patients receiving Cimzia 200 mg every 2 weeks + MTX, clinical responses were seen in some patients within one (22.9%) to two (33.5%) weeks after initiation of therapy.
The percentage of patients achieving ACR20 response by visit for Study RA-I is shown in Figure 1. Among patients receiving Cimzia 200 mg every 2 weeks + MTX, clinical responses were seen in some patients within one (22.9%) to two (33.5%) weeks after initiation of therapy. The safety and efficacy of 400 mg Cimzia administered every 4 weeks in combination with MTX were evaluated Study RA-IV. The primary endpoint of this study was achieved; the proportion of subjects who achieved an ACR20 response at Week 24 was significantly greater in the Cimzia 400 mg + MTX group compared to the placebo + MTX group (45.9% compared to 22.9%, p < 0.001).
The safety and efficacy of 400 mg Cimzia administered every 4 weeks in combination with MTX were evaluated Study RA-IV. The primary endpoint of this study was achieved; the proportion of subjects who achieved an ACR20 response at Week 24 was significantly greater in the Cimzia 400 mg + MTX group compared to the placebo + MTX group (45.9% compared to 22.9%, p < 0.001).
 Of the 783 patients initially randomized to active treatment in RA-I, 508 completed 52 weeks of placebo controlled treatment and entered the open label extension study. Sustained inhibition of progression of structural damage was demonstrated in a subset of 449 of these patients who completed at least 2 years of treatment with Cimzia (RA-I and open label extension study) and had evaluable data at the 2 year timepoint. This was not a preplanned analysis. Linear imputation was used for missing data. There were 177 patients in the control group, including 136 withdrawers (subjects who received placebo + MTX for 12 weeks and failed to achieve an ACR20 response at Week 12, confirmed at Week 14 who then participated in the open label extension study from Week 16) and 41 completers (subjects who received placebo + MTX for 52 weeks before participating in the open label extension study). The efficacy of Cimzia on radiographic endpoints has not been established in patients who are unable to tolerate MTX therapy.
Of the 783 patients initially randomized to active treatment in RA-I, 508 completed 52 weeks of placebo controlled treatment and entered the open label extension study. Sustained inhibition of progression of structural damage was demonstrated in a subset of 449 of these patients who completed at least 2 years of treatment with Cimzia (RA-I and open label extension study) and had evaluable data at the 2 year timepoint. This was not a preplanned analysis. Linear imputation was used for missing data. There were 177 patients in the control group, including 136 withdrawers (subjects who received placebo + MTX for 12 weeks and failed to achieve an ACR20 response at Week 12, confirmed at Week 14 who then participated in the open label extension study from Week 16) and 41 completers (subjects who received placebo + MTX for 52 weeks before participating in the open label extension study). The efficacy of Cimzia on radiographic endpoints has not been established in patients who are unable to tolerate MTX therapy.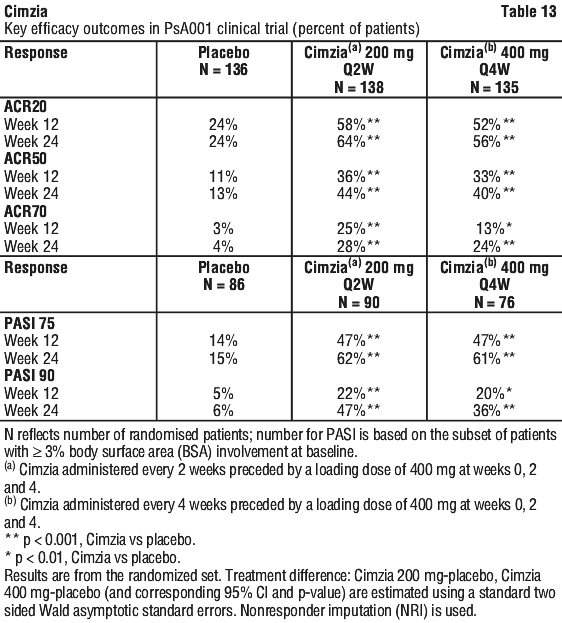
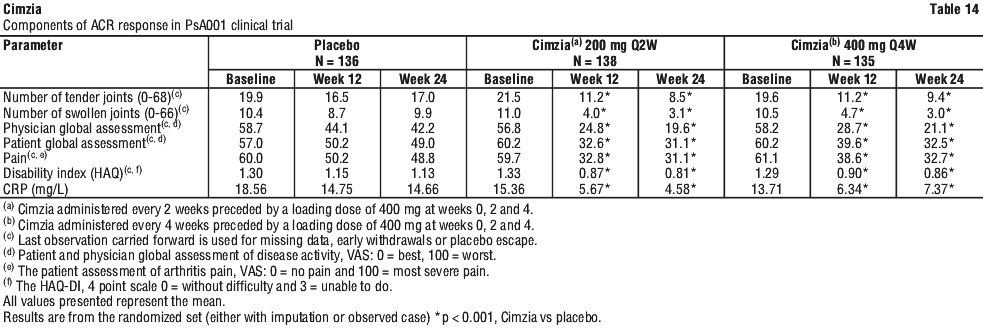 The percentage of ACR20 responders was clinically relevant and significantly higher for the Cimzia 200 mg every 2 weeks and Cimzia 400 mg every 4 weeks treatment groups compared to placebo group at every visit after baseline through week 24 (p ≤ 0.001 at each visit).
The percentage of ACR20 responders was clinically relevant and significantly higher for the Cimzia 200 mg every 2 weeks and Cimzia 400 mg every 4 weeks treatment groups compared to placebo group at every visit after baseline through week 24 (p ≤ 0.001 at each visit).
 In the AS subpopulation, the percentage of ASAS20 responders was clinically relevant and significantly higher for the Cimzia 200 mg every 2 weeks and Cimzia 400 mg every 4 weeks treatment groups compared to placebo group at every visit after baseline through Week 24 (p < 0.05 at each visit).
In the AS subpopulation, the percentage of ASAS20 responders was clinically relevant and significantly higher for the Cimzia 200 mg every 2 weeks and Cimzia 400 mg every 4 weeks treatment groups compared to placebo group at every visit after baseline through Week 24 (p < 0.05 at each visit). Results for Part B are presented in Tables 17 and 18.
Results for Part B are presented in Tables 17 and 18.

 The results of the components of the ASAS40 response criteria are shown in Table 20.
The results of the components of the ASAS40 response criteria are shown in Table 20. Signs of inflammation were assessed by MRI at Week 12 and expressed as change from baseline in SPARCC (Spondyloarthritis Research Consortium of Canada) score for sacroiliac joints. At Week 12, significant inhibition of inflammation in sacroiliac joints was demonstrated by a reduction of inflammation in Cimzia-treated patients (-4.3) vs placebo-treated patients (0.3) as compared to baseline. Patients treated with Cimzia achieved a greater reduction in nocturnal spinal pain (NRS) compared to placebo-treated patients (reduction compared to baseline -4.0 vs -2.1) at Week 52.
Signs of inflammation were assessed by MRI at Week 12 and expressed as change from baseline in SPARCC (Spondyloarthritis Research Consortium of Canada) score for sacroiliac joints. At Week 12, significant inhibition of inflammation in sacroiliac joints was demonstrated by a reduction of inflammation in Cimzia-treated patients (-4.3) vs placebo-treated patients (0.3) as compared to baseline. Patients treated with Cimzia achieved a greater reduction in nocturnal spinal pain (NRS) compared to placebo-treated patients (reduction compared to baseline -4.0 vs -2.1) at Week 52. The key results of the CIMPACT trial are presented in Table 22.
The key results of the CIMPACT trial are presented in Table 22. In the CIMPACT study, at Week 12, the Cimzia 400 mg every 2 weeks dosing schedule demonstrated superiority against Enbrel 50 mg twice weekly in PASI 75 response rate (66.7% and 53.3% respectively, p < 0.05). The Cimzia 200 mg every 2 weeks dosing schedule demonstrated noninferiority against Enbrel 50 mg twice weekly in PASI 75 response rate (61.3%, difference between Enbrel and Cimzia 200 mg every 2 weeks was 8.0%, 95% CI: -2.9, 18.9) based on a pre-specified noninferiority margin of 10%.
In the CIMPACT study, at Week 12, the Cimzia 400 mg every 2 weeks dosing schedule demonstrated superiority against Enbrel 50 mg twice weekly in PASI 75 response rate (66.7% and 53.3% respectively, p < 0.05). The Cimzia 200 mg every 2 weeks dosing schedule demonstrated noninferiority against Enbrel 50 mg twice weekly in PASI 75 response rate (61.3%, difference between Enbrel and Cimzia 200 mg every 2 weeks was 8.0%, 95% CI: -2.9, 18.9) based on a pre-specified noninferiority margin of 10%.

