1 Name of Medicine
Columvi 2.5 mg concentrate for solution for infusion.
Columvi 10 mg concentrate for solution for infusion.
2 Qualitative and Quantitative Composition
Columvi 2.5 mg concentrated injection.
Each vial of 2.5 mL contains 2.5 mg of glofitamab at a concentration of 1 mg/mL.
Columvi 10 mg concentrated injection.
Each vial of 10 mL contains 10 mg of glofitamab at a concentration of 1 mg/mL.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Concentrated injection for intravenous infusion.
Columvi is a preservative-free, colourless, clear solution supplied in single-dose vials.
4.1 Therapeutic Indications
Columvi monotherapy with obinutuzumab pretreatment has provisional approval for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) after two or more lines of systemic therapy. Columvi is not indicated for the treatment of patients with primary central nervous system lymphoma.
The decision to approve this indication has been made on the basis of complete response and the overall response rate from an uncontrolled, open label phase I/II study. Continued approval of this indication depends on verification and description of benefit in confirmatory trials.
Columvi in combination with gemcitabine and oxaliplatin with obinutuzumab pretreatment is indicated for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma not otherwise specified (DLBCL NOS) who are not candidates for autologous stem cell transplant (ASCT). Columvi is not indicated for the treatment of patients with primary central nervous system lymphoma.
4.2 Dose and Method of Administration
General.
Substitution by any other biological medicinal product requires the consent of the prescribing physician.
Columvi therapy should only be administered under the supervision of a healthcare professional experienced in the treatment of cancer patients and who has access to appropriate medical support to manage severe reactions associated with cytokine release syndrome (CRS).
At least 1 dose of tocilizumab for use in the event of CRS must be available prior to Columvi infusion at cycles 1 and 2. Access to an additional dose of tocilizumab within 8 hours of use of the previous tocilizumab dose must be ensured. See Section 4.4 Special Warnings and Precautions for Use.
Pre-treatment with obinutuzumab. All patients must receive a single 1000 mg dose of obinutuzumab on cycle 1 day 1 (7 days prior to initiation of Columvi treatment); see Table 2, Table 3 and Delayed or missed doses. This is to deplete circulating and lymphoid tissue B cells and thereby reduce the risk of CRS.
Obinutuzumab should be administered as an intravenous infusion at 50 mg/h. The rate of infusion can be escalated in 50 mg/h increments every 30 minutes to a maximum of 400 mg/h.
Refer to the obinutuzumab Product Information for complete information on premedication, preparation, administration, and management of adverse reactions of obinutuzumab.
Premedication and prophylactic medications. Cytokine release syndrome prophylaxis.
Columvi should be administered to well-hydrated patients. Premedication to reduce the risk of CRS (see Section 4.4 Special Warnings and Precautions for Use) is outlined in Table 1.
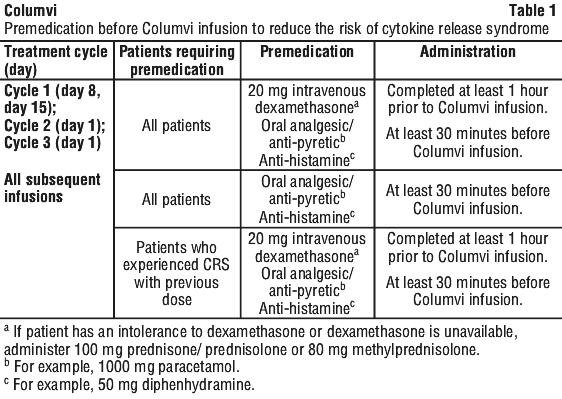
Infection prophylaxis.
Prophylaxis is recommended to reduce the risk of infection (see Section 4.4 Special Warnings and Precautions for Use).
Consider prophylaxis for cytomegalovirus (CMV), herpes, Pneumocystis jirovecii pneumonia, and other opportunistic infections in patients at increased risk (see Section 4.8 Adverse Effects (Undesirable Effects)).
Recommended dosage.
Columvi dosing begins with a step-up dosing schedule (which is designed to decrease the risk of CRS), leading to the recommended dose of 30 mg.
Columvi monotherapy dose step-up schedule.
Columvi must be administered as an intravenous infusion according to the dose step-up schedule leading to the recommended dosage of 30 mg (as shown in Table 2), after completion of pre-treatment with obinutuzumab on cycle 1 day 1. Each cycle is 21 days.

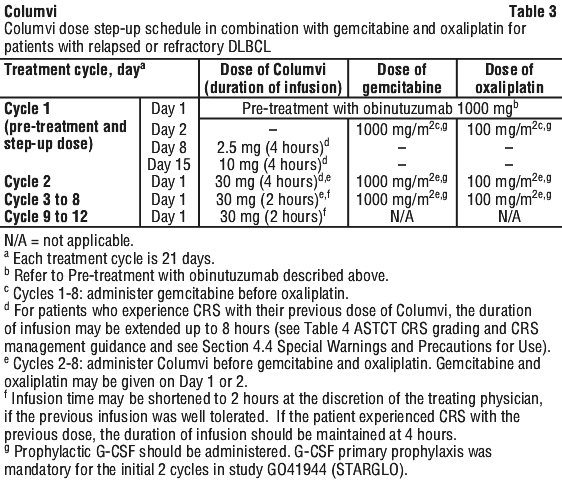
Columvi dose step-up schedule in combination with gemcitabine and oxaliplatin.
Columvi must be administered as an intravenous infusion according to the dose step-up schedule leading to the recommended dosage of 30 mg (as shown in Table 3), after completion of pre-treatment with obinutuzumab on cycle 1 day 1. Administer Columvi in combination with gemcitabine and oxaliplatin at cycles 1-8 and as monotherapy at cycles 9-12. Each cycle is 21 days.
Monitoring after infusion.
All patients must be monitored for signs and symptoms of potential CRS during infusion and for at least 10 hours after completion of the infusion of the first Columvi dose (2.5 mg on cycle 1 day 8).
Patients who experienced grade ≥ 2 CRS with their previous infusion should be monitored after completion of the infusion (see Table 4).
All patients should be monitored for signs and symptoms of CRS and/or neurologic toxicity including immune effector cell-associated neurotoxicity syndrome (ICANS) following Columvi administration. All patients must be counselled on the risk, signs, and symptoms of CRS and/or neurologic toxicity including ICANS and advised to contact the healthcare provider immediately should they experience signs and symptoms of CRS and/or neurologic toxicity including ICANS at any time.
Duration of treatment.
Treatment with Columvi monotherapy is recommended for a maximum of 12 cycles or until disease progression or unmanageable toxicity, whichever occurs first.
Treatment with Columvi in combination with gemcitabine and oxaliplatin is recommended for 8 cycles, followed by 4 cycles of Columvi monotherapy for a maximum of 12 cycles of Columvi in total or until disease progression or unmanageable toxicity, whichever occurs first.
Delayed or missed doses.
During step-up dosing (weekly dosing).
Following pre-treatment with obinutuzumab, if the Columvi 2.5 mg dose is delayed by more than 1 week, then repeat pre-treatment with obinutuzumab.
Following Columvi 2.5 mg dose or 10 mg dose, if there is a Columvi treatment-free interval of 2 weeks to 6 weeks, then repeat the last tolerated Columvi dose and resume the planned step-up dosing.
Following Columvi 2.5 mg dose or 10 mg dose, if there is a Columvi treatment-free interval of more than 6 weeks, then repeat pre-treatment with obinutuzumab and ColumviI step-up dosing (see cycle 1 in Table 2 and Table 3).
After cycle 2 (30 mg dose).
If there is a Columvi treatment-free interval of more than 6 weeks between cycles, then repeat pre-treatment with obinutuzumab and Columvi step-up dosing (see cycle 1 in Table 2 and Table 3) and then resume the planned treatment cycle (30 mg dose).
Preparation and administration of Columvi.
Preparation.
Columvi must be diluted by a healthcare professional using aseptic technique, prior to intravenous administration. For instructions on dilution of the medicine before administration, see Section 6.6 Special Precautions for Disposal.
Administration.
Columvi must be administered as an intravenous infusion through a dedicated infusion line via syringe or bag infusion over a maximum of 8 hours.
Columvi must not be administered as an intravenous push or bolus.
Columvi must not be mixed with other drugs.
Dose modifications.
No dose reductions of Columvi are recommended.
Management of cytokine release syndrome (CRS).
CRS should be identified based on the clinical presentation (see Section 4.4 Special Warnings and Precautions for Use). Patients should be evaluated for other causes of fever, hypoxia, and hypotension, such as infections or sepsis. If CRS is suspected, it should be managed according to the CRS management recommendations based on American Society for Transplantation and Cellular Therapy [ASTCT] consensus grading in Table 4.

Management of neurologic toxicity including immune effector cell-associated neurotoxicity syndrome (ICANS).
Neurologic toxicity including ICANS should be identified based on the clinical presentation (see Section 4.4 Special Warnings and Precautions for Use). At the first sign of neurologic toxicity, including ICANS, based on the type and severity of neurologic toxicity consider supportive therapy, neurology evaluation, and withholding Columvi per Table 5. Rule out other causes of neurologic symptoms. If ICANS is suspected, it should be managed according to the recommendations in Table 5.

Special populations.
Elderly.
No dose adjustment of Columvi is required in patients ≥ 65 years of age (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetics Properties, Pharmacokinetics in special populations).
Renal impairment.
No dose adjustment of Columvi is required in patients with mild or moderate renal impairment (CrCl 30 to < 90 mL/min). Columvi has not been studied in patients with severe renal impairment (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetics Properties, Pharmacokinetics in special populations).
Hepatic impairment.
No dose adjustment is required in patients with mild hepatic impairment (total bilirubin > upper limit of normal [ULN] to ≤ 1.5 x ULN or aspartate transaminase [AST] > ULN). No specific studies in patients with moderate or severe hepatic impairment have been conducted with Columvi (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetics Properties, Pharmacokinetics in special populations).
Paediatric populations.
The safety and efficacy of Columvi in paediatric patients have not been established.4.3 Contraindications
Columvi is contraindicated in patients with a known hypersensitivity to glofitamab or any of the excipients.
Refer to obinutuzumab-specific contraindications in the obinutuzumab Product Information.
4.4 Special Warnings and Precautions for Use
Refer to obinutuzumab-specific warnings and precautions in the obinutuzumab Product Information.
In order to improve traceability of biological medicinal products, the trade name and the batch number of the administered product should be clearly recorded (or stated) in the patient file.
Cytokine release syndrome.
CRS, including life-threatening reactions, has been reported in patients receiving Columvi.
The most common manifestations of CRS were pyrexia, tachycardia, hypotension, chills, and hypoxia. Infusion-related reactions may be clinically indistinguishable from manifestations of CRS.
To reduce the occurrence of CRS, patients must be pre-treated with obinutuzumab, 7 days prior to initiation of Columvi, and should be premedicated with an anti-pyretic, anti-histamine, and a glucocorticoid (see Section 4.2 Dose and Method of Administration).
At least 1 dose of tocilizumab for use in the event of CRS must be available prior to Columvi infusion at cycles 1 and 2. Access to an additional dose of tocilizumab within 8 hours of use of the previous tocilizumab dose must be ensured.
Patients must be monitored during all Columvi infusions and for at least 10 hours after completion of the first infusion.
For complete information on monitoring, see Section 4.2 Dose and Method of Administration. The prescriber must counsel patients to seek immediate medical attention should signs or symptoms of CRS occur at any time.
Patients should be evaluated for other causes of fever, hypoxia, and hypotension, such as infections or sepsis. CRS should be managed based on the patient's clinical presentation and according to the CRS management guidance provided in Table 4 (see Section 4.2 Dose and Method of Administration).
Neurologic toxicity including immune effector cell-associated neurotoxicity syndrome (ICANS).
Neurologic toxicity including ICANS has been reported in patients receiving Columvi, including serious and fatal reactions. Manifestations of ICANS included somnolence, cognitive disorder, confusional state, delirium, and disorientation. The majority of cases of ICANS occurred during cycle 1 of Columvi treatment, however, some events occurred at later cycles (see Section 4.8 Adverse Effects (Undesirable Effects), Description of selected adverse drug reactions from clinical trials).
Patients should be monitored for signs and symptoms of neurologic toxicity including ICANS following Columvi administration. The prescriber must counsel patients to seek immediate medical attention should signs or symptoms occur at any time.
At the first signs or symptoms of ICANS, manage according to the ICANS guidance provided in Table 5. Treatment with Columvi should be withheld or discontinued as recommended (see Section 4.2 Dose and Method of Administration).
Patient card.
The prescriber must inform the patient of the risk of CRS and ICANS and the signs and symptoms of CRS and ICANS. Patients must be instructed to seek immediate medical attention if they experience signs and symptoms of CRS and ICANS. Patients should be provided with the Patient card and instructed to carry the card at all times. This card describes symptoms of CRS and ICANS which, if experienced, should prompt the patient to seek immediate medical attention.
Serious infections.
Serious infections (including opportunistic infections) have occurred in patients treated with Columvi (see Section 4.8 Adverse Effects (Undesirable Effects)).
Columvi must not be administered to patients with an active infection. Caution should be exercised when considering the use of Columvi in patients with a history of chronic or recurrent infection, those with underlying conditions that may predispose them to infections, or those who have had significant prior immunosuppressive treatment. Administer prophylactic antimicrobials, as appropriate. Viral reactivation, e.g. Hepatitis B virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure and death, can occur in patients treated with drugs directed against B cells, the frequency of these events in unknown with Columvi. Patients should be monitored before and during Columvi treatment for the emergence of possible bacterial, fungal, and new or reactivated viral infections and treated appropriately.
Columvi should be temporarily withheld in the presence of an active infection until the infection has resolved. Patients should be instructed to seek medical advice if signs and symptoms suggestive of an infection occur.
Febrile neutropenia has been reported during treatment with Columvi. Patients with febrile neutropenia should be evaluated for infection and treated promptly.
Tumour flare.
Tumour flare has been reported in patients receiving Columvi. Manifestations included localised pain and swelling (see Section 4.8 Adverse Effects (Undesirable Effects)).
Consistent with the mechanism of action of Columvi, tumour flare is likely due to the influx of T-cells into tumour sites following Columvi administration and may mimic progression of disease. Tumour flare does not imply treatment failure or represent tumour progression.
Specific risk factors for tumour flare have not been identified, however, there is a heightened risk of compromise and morbidity due to mass effect secondary to tumour flare in patients with bulky tumours located in close proximity to airways and/or a vital organ. Monitoring and evaluation of tumour flare at critical anatomical sites is recommended in patients treated with Columvi and managed as clinically indicated.
Tumour lysis syndrome (TLS).
TLS has been reported in patients receiving Columvi (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients with high tumour burden, rapidly proliferative tumours, renal dysfunction, or dehydration are at greater risk of TLS.
Patients at risk should be monitored closely by appropriate clinical and laboratory tests for electrolyte status, hydration, and renal function. Appropriate prophylactic measures with anti-hyperuricemics (e.g. allopurinol or rasburicase) and adequate hydration should be considered prior to Columvi infusion.
Management of TLS may include aggressive hydration, correction of electrolyte abnormalities, anti-hyperuricemic therapy, and supportive care.
Immunisation.
The safety of immunisation with live vaccines during or following Columvi therapy has not been studied. Immunisation with live vaccines is not recommended during Columvi therapy.
Primary central nervous system (CNS) lymphoma.
There is no experience of use of Columvi in patients with primary CNS lymphoma. Therefore, the risk/benefit of Columvi has not been established in this population.
Use in hepatic impairment.
No dose adjustment is required in patients with mild hepatic impairment (total bilirubin > ULN to ≤ 1.5 × ULN or AST > ULN) based on population pharmacokinetic analysis. The safety and efficacy of Columvi in patients with moderate or severe hepatic impairment has not been studied (See Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetics Properties, Pharmacokinetics in special populations).
Use in renal impairment.
No dose adjustment is required in patients with mild or moderate renal impairment (CrCl 30 to < 90 mL/min) based on population pharmacokinetic analysis. The safety and efficacy of Columvi in patients with severe renal impairment has not been studied (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetics Properties, Pharmacokinetics in special populations).
Use in the elderly.
No differences in safety or efficacy of Columvi were observed between patients ≥ 65 years of age and those under 65 years. No dose adjustment of Columvi is required in patients ≥ 65 years of age (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetics Properties, Pharmacokinetics in special populations).
Paediatric use.
The safety and efficacy of Columvi in paediatric patients have not been established.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No clinical drug-drug interaction studies have been performed.
No interaction studies have been performed. Though the initial release of cytokines associated with the start of Columvi treatment could suppress CYP450 enzymes, these changes would be transient as cytokine release is only observed with the first doses of Columvi. The highest drug-drug interaction risk is during the period of one week following each of the first 2 doses of Columvi (i.e. cycle 1 day 8 and 15) in patients who are receiving concomitant CYP450 substrates with a narrow therapeutic index (e.g. warfarin, cyclosporine). On initiation of Columvi therapy, patients being treated with CYP450 substrates with a narrow therapeutic index should be monitored.
The pharmacokinetics of glofitamab are not affected by co-administration with gemcitabine or oxaliplatin.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No fertility studies in animals have been performed to evaluate the effect of Columvi.
(Category C)
Female patients of reproductive potential must be advised to avoid pregnancy while receiving Columvi. There are no available data on the use of Columvi in pregnant women. Glofitamab is an immunoglobulin G (IgG). IgG is known to cross the placenta. Based on its mechanism of action, glofitamab is likely to cause fetal B-cell depletion when administered to a pregnant woman, posing a risk of opportunistic infections in the neonate. Glofitamab-induced cytokine release may also pose a risk for embryofetal loss. The risk of malformations is considered to be low.
Postponing vaccination with live or live-attenuated vaccines is recommended for neonates and infants who have been exposed to glofitamab in utero until B-cell levels have recovered. Female patients receiving Columvi should be advised of the potential harm to the fetus. Female patients should be advised to contact the treating physician, should pregnancy occur.
Contraception.
Female patients of reproductive potential must use highly effective contraceptive methods during treatment and for at least 2 months following the last dose of Columvi. Medications co-administered with Columvi may have different recommendations regarding fertility, pregnancy and lactation. Please refer to their respective Product Information's, if needed.
Labour and delivery.
The safe use of Columvi during labour and delivery has not been established.
It is not known whether glofitamab is excreted in human milk. No studies have been conducted to assess the impact of glofitamab on milk production or its presence in human milk. Human IgG is known to be present in human milk. The potential for absorption of glofitamab and the potential for adverse reactions in the nursing infant is unknown. Women should be advised to discontinue breastfeeding during treatment with Columvi and for 2 months after the last dose of Columvi.4.7 Effects on Ability to Drive and Use Machines
Columvi has no or negligible influence on the ability to drive and use machines. Patients experiencing symptoms of neurologic toxicity including ICANS and/or CRS (pyrexia, tachycardia, hypotension, chills, hypoxia) should be advised not to drive or use machines until symptoms resolve.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
Approximately 1530 patients with relapsed or refractory non-Hodgkin's lymphoma have received Columvi in the clinical development program of Columvi.
The adverse drug reactions described below were identified from clinical studies in patients with relapsed or refractory DLBCL who were treated with Columvi at the recommended dose as monotherapy (N = 145) or in combination with gemcitabine and oxaliplatin (N = 172).
Tabulated summary of adverse drug reactions from clinical trials.
Adverse drug reactions from clinical trials (see Table 6, Table 7) are listed by MedDRA system organ class. The corresponding frequency category for each adverse drug reaction is based on the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000).
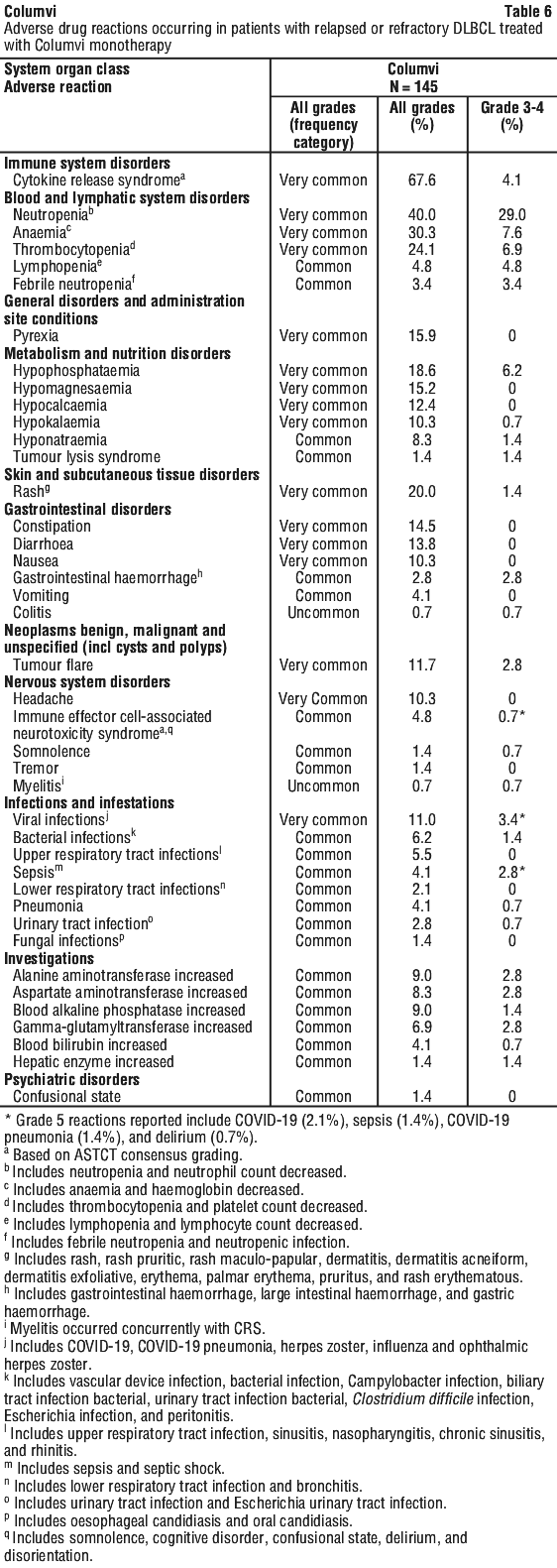
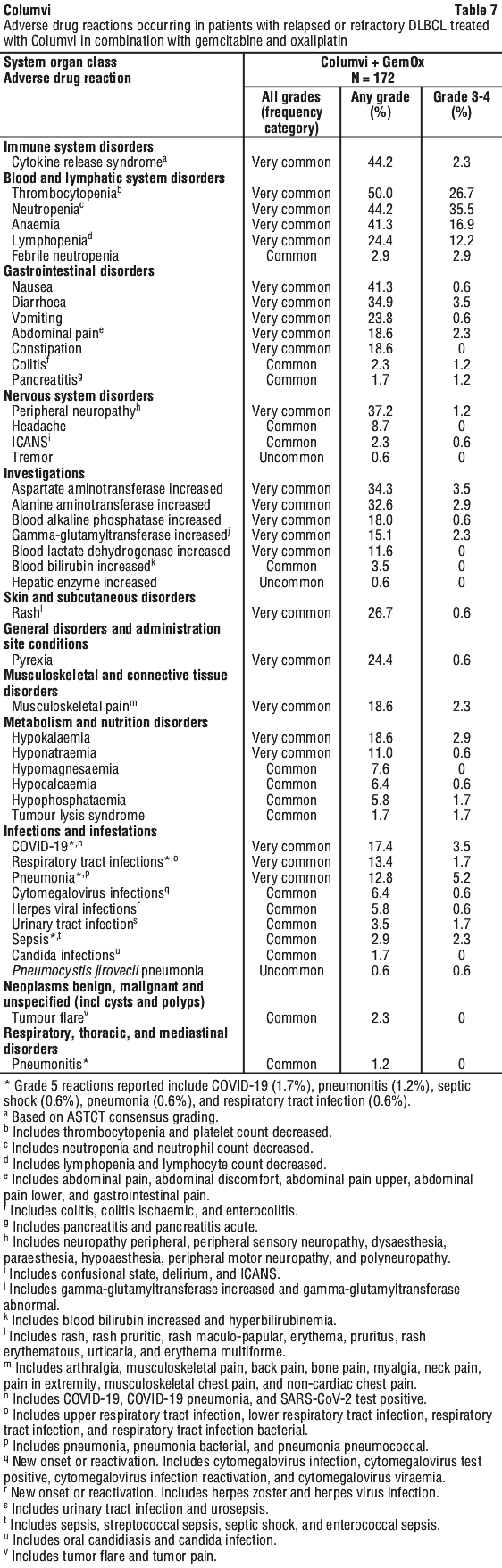
Description of selected adverse drug reactions from clinical trials.
The descriptions below reflect information for significant adverse reactions for Columvi monotherapy and/or combination therapy. Details for the significant adverse reactions for Columvi when given in combination are presented if clinically relevant differences were noted in comparison to Columvi monotherapy.
Cytokine release syndrome. Columvi monotherapy.
Any grade CRS (by ASTCT criteria) occurred in 67.6% of patients who received Columvi monotherapy, with grade 1 CRS reported in 50.3% of patients, grade 2 CRS in 13.1% of patients, grade 3 CRS in 2.8% of patients, and grade 4 CRS in 1.4% of patients. There were no fatal cases of CRS. CRS resolved in all patients except one. One patient discontinued Columvi due to CRS.
In patients with CRS, the most common manifestations of CRS included pyrexia (99.0%), tachycardia (25.5%), hypotension (23.5%), chills (14.3%), and hypoxia (12.2%). Grade 3 or higher events associated with CRS included hypotension (3.1%), hypoxia (3.1%), pyrexia (2.0%), and tachycardia (2.0%).
CRS of any grade occurred in 54.5% of patients following the 2.5 mg dose of Columvi at cycle 1 day 8 with median time to onset (from the start of infusion) of 12.6 hours (range: 5.2 to 50.8 hours); in 33.3% of patients following the 10 mg dose at cycle 1 day 15 with median time to onset of 26.8 hours (range: 6.7 to 125.0 hours); and in 26.8% of patients following the 30 mg dose at cycle 2 day 1 with median time to onset of 28.2 hours (range: 15.0 to 44.2 hours). CRS was reported in 0.9% of patients at cycle 3 and in 2% of patients beyond cycle 3.
Grade ≥ 2 CRS occurred in 12.4% of patients following the first Columvi dose (2.5 mg), with median time to onset of 9.7 hours (range: 5.2 to 19.1 hours) and median duration of 50.4 hours (range: 6.5 to 316.7 hours). Following Columvi 10 mg dose at cycle 1 day 15, the incidence of grade ≥ 2 CRS decreased to 5.2% of patients, with median time to onset of 26.2 hours (range: 6.7 to 144.2 hours) and median duration of 30.9 hours (range: 3.7 to 227.2 hours). Grade ≥ 2 CRS following Columvi 30 mg dose at cycle 2 day 1 occurred in one patient (0.8%) with time to onset of 15.0 hours and duration of 44.8 hours. No grade ≥ 2 CRS was reported beyond cycle 2.
Among the 25 patients who experienced grade 2 or higher CRS after Columvi, 22 (88%) received tocilizumab, 15 (60%) received corticosteroids, and 14 (56%) received both tocilizumab and corticosteroids. Ten patients (40%) received oxygen. All 6 patients (24.0%) with grade 3-4 CRS received a single vasopressor.
In patients who received dexamethasone premedication (n = 39) versus another glucocorticoid premedication (n = 106), CRS of any grade occurred in 48.7% vs. 56.6% of patients; grade 1 CRS in 38.5% vs. 43.4% of patients; grade 2 CRS in 7.7% vs. 9.4% of patients; grade 3 CRS in 2.6% vs. 1.9% of patients; and grade 4 CRS in 0% vs. 1.9% of patients after the 2.5 mg dose of Columvi at cycle 1 day 8.
Columvi in combination with gemcitabine and oxaliplatin.
Any grade CRS (by ASTCT criteria) occurred in 44.2% of patients who received Columvi with gemcitabine and oxaliplatin, with grade 1 CRS reported in 31.4% of patients, grade 2 CRS in 10.5% of patients and grade 3 CRS in 2.3% of patients. There were no grade 4 or fatal cases of CRS. CRS resolved in all patients except one. One patient discontinued Columvi due to CRS.
In patients with CRS, the most common manifestations of CRS included pyrexia (98.7%), hypotension (22.4%), chills (17.1%) and hypoxia (14.5%). Grade 3 or higher events associated with CRS included hypotension (6.6%), hypoxia (5.3%), pyrexia (3.9%), chills (1.3%), and diarrhoea (1.3%).
CRS of any grade occurred in 34.9% of patients following the 2.5 mg dose of Columvi at cycle 1 day 8 with median time to onset (from the start of infusion) of 12.6 hours (range: 4.4 to 54.7 hours); in 14.4% of patients following the 10 mg dose at cycle 1 day 15 with median time to onset of 22.8 hours (range: 7.4 to 81.2 hours); and in 9.3% of patients following the 30 mg dose at cycle 2 day 1 with median time to onset of 23.5 hours (range: 14.7 to 33.4 hours). CRS was reported in 6.7% of patients at cycle 3 and in 11.0% of patients beyond cycle 3.
Grade ≥ 2 CRS occurred in 10.5% of patients following the first Columvi dose (2.5 mg), with median time to onset of 12.0 hours (range: 4.4 to 30.5 hours) and median duration of 42.3 hours (range: 3.5 to 143.7 hours). Following Columvi 10 mg dose at cycle 1 day 15, the incidence of grade ≥ 2 CRS decreased to 1.8% of patients, with median time to onset of 22.3 hours (range: 7.4 to 22.8 hours) and median duration of 37.0 hours (range: 34.8 to 248.5 hours). There were no grade ≥ 2 CRS events following Columvi 30 mg dose at cycle 2 day 1. Three patients (2%) had grade ≥ 2 CRS beyond cycle 2 (all grade 2 events).
Among the 22 patients who experienced grade 2 or higher CRS after Columvi, 16 (72.7%) received tocilizumab, 15 (68.2%) received corticosteroids, and 12 (54.5%) received both tocilizumab and corticosteroids. Eleven (50.0%) received oxygen. All 4 patients (18.2%) with grade 3 CRS received a single vasopressor.
Neurologic toxicity, including immune effector cell-associated neurotoxicity syndrome (ICANS). Columvi monotherapy.
Neurologic toxicity was reported in 40.0% of patients who received Columvi monotherapy. The most frequent neurologic toxicities of any grade were headache (10.3%), dizziness (5.5%), anxiety (4.1%), and paraesthesia (2.8%). Grade 3 or higher neurologic adverse reactions occurred in 2.1% of patients and included somnolence, delirium, and myelitis.
Seven patients (4.8%) experienced ICANS: 5 patients (3.4%) experienced grade 1 events (2 patients with confusional state, and 1 patient each with somnolence, cognitive disorder, and disorientation), and 1 patient (0.7%) experienced grade 3 somnolence. One patient (0.7%) experienced a fatal (grade 5) event of delirium.
The median time to onset of the first ICANS event was 8 days (range: 1 to 106 days) from the first Columvi dose. The median time to resolution of ICANS was 1.5 days (range: 1 to 8 days). Of the 7 patients with ICANS, the onset of ICANS was concurrent with CRS in 5 patients and was non-concurrent with CRS in 2 patients.
Columvi in combination with gemcitabine and oxaliplatin.
Neurologic toxicity was reported in 59.3% of patients who received Columvi with gemcitabine and oxaliplatin. The most frequent neurologic toxicities of any grade were peripheral neuropathy (37.2%), insomnia (11.6%), headache (8.7%), dizziness (7.0%) and dysgeusia (5.2%). Grade 3 or higher neurologic adverse reactions occurred in 5.8% of patients and included peripheral sensory neuropathy, syncope, extrapyramidal disorder, confusional state, delirium, herpes zoster, meningitis aseptic, subdural haematoma, and cerebral haemorrhage.
Four patients (2.3%) experienced ICANS: 1 patient (0.6%) experienced a grade 1 event (confusional state), 2 patients (1.2%) experienced a grade 2 event (1 patient with confusional state, and 1 patient with ICANS) and 1 patient (0.6%) experienced a grade 3 (delirium).
The onset of the 4 events of ICANS was concurrent with CRS. The median time to resolution of ICANS was 1.5 days (range: 1 to 4 days).
Serious infections. Serious infections were reported in 15.9% of patients who received Columvi monotherapy. The most frequent serious infections reported in ≥ 2% of patients were sepsis (4.1%), COVID-19 (3.4%), and COVID-19 pneumonia (2.8%). Infection-related deaths were reported in 4.8% of patients (due to sepsis, COVID-19 pneumonia, and COVID-19). Four patients (2.8%) experienced serious infections concurrently with grade 3-4 neutropenia.
Serious infections were reported in 22.7% of patients who received Columvi with gemcitabine and oxaliplatin. The most frequent serious infections reported in ≥ 2% patients were pneumonia (5.8%), COVID-19 (4.7%), and lower respiratory tract infection (2.9%). Infection-related deaths were reported in 3.5% of patients (due to COVID-19, pneumonia, respiratory tract infection, and septic shock). One patient (0.6%) experienced a serious infection (pneumonia) concurrently with grade 3 neutropenia.
Pneumonitis. Pneumonitis adverse events (excluding pneumonia of infectious etiology) were reported in 2 patients (1.2%) who received Columvi with gemcitabine and oxaliplatin, both of which were fatal events. The median time to onset of pneumonitis from the first glofitamab dose was 168 days (range: 102 to 255 days).
Colitis. Colitis (grade 4) was reported in 1 patient who received Columvi monotherapy, with time to onset from the first Columvi dose of 104 days.
Colitis adverse events (excluding infectious etiology) were reported in 4 patients (2.3%) who received Columvi with gemcitabine and oxaliplatin. Two patients (1.2%) had grade 3 events. The median time to onset of colitis from the first glofitamab dose was 154 days (range: 115 to 187 days).
Opportunistic infections. Cytomegalovirus (CMV) events were reported in 6/467 patients (1.3%) who received Columvi monotherapy in study NP30179, with 1 patient (0.2%) experiencing grade 3 CMV chorioretinitis. Pneumocystis jirovecii pneumonia was reported in 4/467 patients (0.9%), 3 of whom (0.6%) had grade 3 events.
CMV events were reported in 11 patients (6.4%) who received Columvi with gemcitabine and oxaliplatin, with 1 patient (0.6%) experiencing a grade 3 CMV viremia. Oral candidiasis was reported in 3 patients (1.7%) all of which were grade 1-2 events. Pneumocystis jirovecii pneumonia (grade 3) was reported in 1 patient (0.6%), the same patient with grade 3 CMV viremia. Borellia meningitis (grade 2) was reported in 1 patient (0.6%).
Neutropenia. Neutropenia (including neutrophil count decreased) was reported in 40.0% of patients and severe neutropenia (grade 3-4) was reported in 29.0% of patients who received Columvi monotherapy. The median time to onset of the first neutropenia event was 29 days (range: 1 to 203 days). Prolonged neutropenia (lasting longer than 30 days) occurred in 11.7% of patients. The majority of patients with neutropenia (79.3%) were treated with G-CSF. Febrile neutropenia was reported in 3.4% of patients.
All patients who were treated with Columvi with gemcitabine and oxaliplatin in study GO41944 (STARGLO) received prophylactic G-CSF for the initial 2 cycles. In this population, neutropenia (including neutrophil count decreased) was reported in 44.2% of patients and severe neutropenia (grade 3−4) was reported in 35.5% of patients. The median time to onset of the first neutropenia event was 58 days (range: 1 to 224 days). Prolonged neutropenia (lasting longer than 30 days) occurred in 8.7% of patients. Febrile neutropenia was reported in 2.9% of patients.
Tumour flare. Tumour flare was reported in 11.7% of patients who received Columvi monotherapy, including grade 2 tumour flare in 4.8% of patients and grade 3 tumour flare in 2.8% of patients. Tumour flare was reported involving lymph nodes in the head and neck presenting with pain, and involving lymph nodes in the thorax with symptoms of breathlessness due to development of pleural effusion. Most tumour flare events (16/17) occurred during cycle 1, and no tumour flare events were reported beyond cycle 2. The median time to onset of tumour flare of any grade was 2 days (range: 1 to 16 days), and the median duration was 3.5 days (range: 1 to 35 days). No patients discontinued Columvi due to tumour flare.
Tumour lysis syndrome (TLS). TLS was reported in 2 patients (1.4%) who received Columvi monotherapy and was grade 3 in severity in both cases. The median time to onset of TLS was 2 days, and the median duration was 4 days (range: 3 to 5 days).
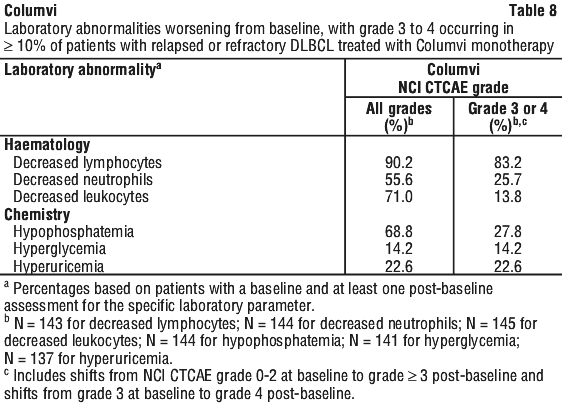
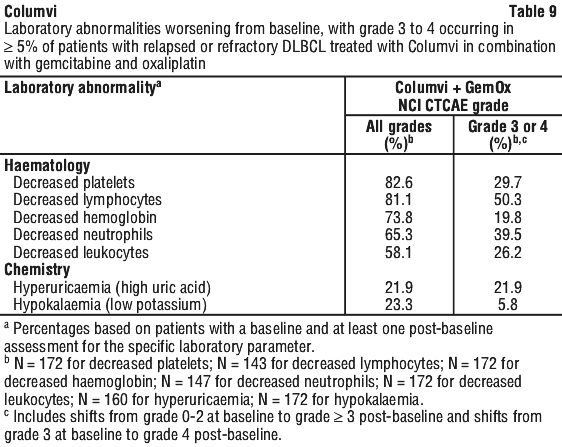
Laboratory abnormalities.
Table 8 summarises treatment-emergent shifts from baseline in laboratory abnormalities with Columvi monotherapy. Table 9 summarises treatment-emergent shifts from baseline in laboratory abnormalities with Columvi in combination with gemcitabine and oxaliplatin.
Reporting of suspected adverse reactions.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no experience with overdosage of Columvi in clinical trials.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: antineoplastic agents, other monoclonal antibodies and antibody drug conjugates, ATC code: L01FX28.
Mechanism of action.
Glofitamab is a bispecific monoclonal antibody that binds bivalently (with high avidity) to CD20 expressed on the surface of B cells and monovalently to CD3 in the T-cell receptor complex expressed on the surface of T-cells. By simultaneous binding to CD20 on the B cell and CD3 on the T-cell, glofitamab mediates the formation of an immunological synapse with subsequent potent T-cell activation and proliferation, secretion of cytokines, and release of cytolytic proteins that results in the lysis of CD20-expressing B-cells.
Pharmacodynamics.
In study NP30179, peripheral B-cell counts were < 70 cell/microL in almost all patients (98.6%) with relapsed and refractory LBCL prior to Columvi treatment initiation and remained low during Columvi treatment.
During cycle 1 (step-up dosing), transient increases in plasma IL-6 levels were observed at 6 hours post-Columvi infusion, which remained elevated at 20 hours post-infusion and returned to baseline prior to the next infusion.
In study GO41944 (STARGLO), peripheral B-cell counts were < 70 cells/microL in the majority of patients (79.4%) with relapsed and refractory DLBCL NOS prior to Columvi treatment initiation and remained low during Columvi treatment.
Cardiac electrophysiology.
In study NP30179, 16/154 patients who received the study treatment experienced a post-baseline QTc value > 450ms. One of these cases was assessed to be of clinical significance by the investigator. No patients discontinued treatment due to QTc prolongation.
Clinical trials.
Relapsed or refractory DLBCL.
Columvi monotherapy. The efficacy of Columvi monotherapy was evaluated in study NP30179, a single arm, open-label multicentre, multi-cohort trial, which included 155 patients with relapsed or refractory DLBCL after at least two prior lines of systemic therapy. The study excluded patients with prior allogeneic haematopoietic stem cell transplant, previous or active central nervous system (CNS) lymphoma, ECOG performance status ≥ 2, creatinine clearance (CrCl) < 50 mL/min, or hepatic transaminases > 3 x ULN.
Following pre-treatment with obinutuzumab at cycle 1 day 1, patients received 2.5 mg of Columvi at cycle 1 day 8, 10 mg of Columvi at cycle 1 day 15, and 30 mg of Columvi at cycle 2 day 1 as per the step-up dosing schedule. patients continued to receive 30 mg of Columvi on day 1 of cycles 3 to 12. Patients received premedication including an anti-pyretic, an anti-histamine and a glucocorticoid (see Section 4.2 Dose and Method of Administration). The duration of each cycle was 21 days. Patients received a median of 5 cycles of Columvi treatment (range: 1 to 12 cycles).
The baseline demographic and disease characteristics were: median age 66 years (range: 21 to 90 years); 65.2% males; 76.8% white, 4.5% Asian, and 1.9% Black or African American; 5.8% Hispanic or Latino; and ECOG performance status of 0 (44.5%) or 1 (54.2%). Most patients (71.0%) had DLBCL not otherwise specified, 18.7% had DLBCL transformed from follicular lymphoma, 6.5% had high-grade B-cell lymphoma (HGBCL), and 3.9% had PMBCL. The median number of prior lines of therapy was 3 (range: 2 to 7), 39.4% of patients received 2 prior lines and 60.6% received 3 or more prior lines of therapy. All patients had received prior chemotherapy and anti-CD20 monoclonal antibody therapy; 33.5% of patients had received prior CAR T-cell therapy, and 18.1% of patients had received autologous stem cell transplant. Most patients (89.7%) had refractory disease, 58.7% patients had primary refractory disease, and 84.5% of patients were refractory to their last prior therapy, and. 88.5% of patients who received prior CAR T-cell therapy were refractory to CAR T-cell therapy. There is limited experience of use of Columvi in patients who received subsequent allogeneic stem cell transplant following Columvi.
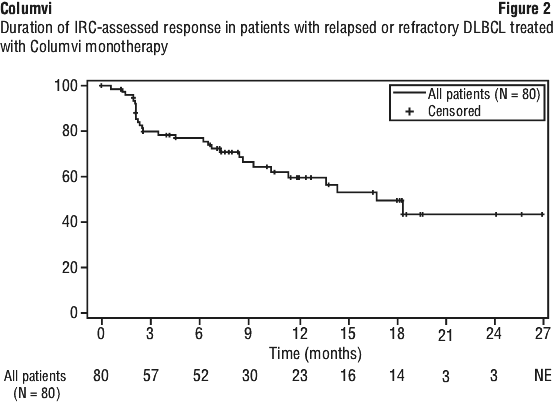
The overall median duration of follow-up was 13.4 months (range: 0 to 28 months). Median duration of follow-up from the date of first response per Independent Review Committee (IRC) assessment was 12.0 months (range: 0 to 27 months).
The primary efficacy outcome measure was complete response (CR) rate as assessed by IRC using 2014 Lugano criteria. The secondary efficacy outcome measures included Investigator (INV)-assessed CR, and overall response rate (ORR), duration of response (DOR), duration of complete response (DOCR), time to first response (TFOR), time to first complete response (TFCR), overall survival (OS), and progression-free survival (PFS), as assessed by IRC and by INV.
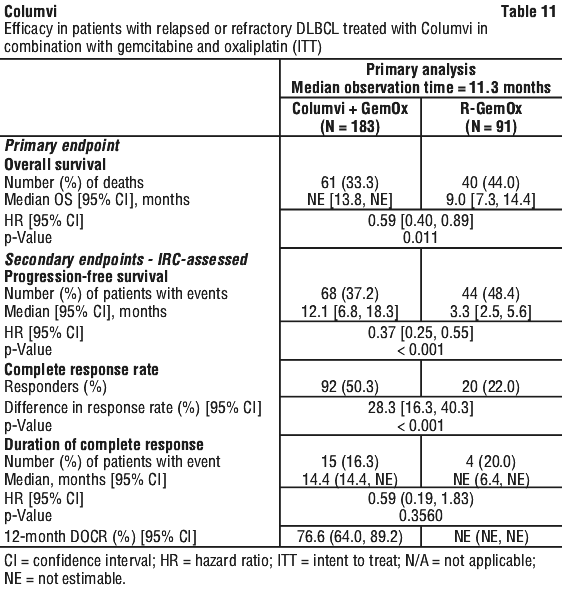
Efficacy results are summarised in Table 10.
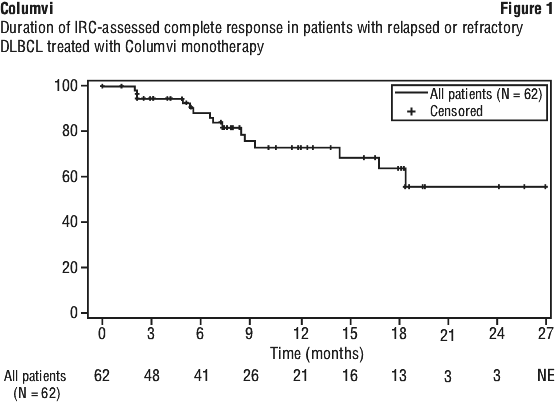
 The efficacy population included a cohort of patients (N = 40) where dexamethasone was mandated as the glucocorticoid premedication. In this cohort, the IRC-assessed ORR was 52.5% (95% CI: 36.1, 68.5) and the CR rate was 47.5% (95% CI: 31.5, 63.9). See Figures 1 and 2.
The efficacy population included a cohort of patients (N = 40) where dexamethasone was mandated as the glucocorticoid premedication. In this cohort, the IRC-assessed ORR was 52.5% (95% CI: 36.1, 68.5) and the CR rate was 47.5% (95% CI: 31.5, 63.9). See Figures 1 and 2.
Patient reported outcomes.
Study NP30179 evaluated patient-reported outcomes of Columvi treatment. Patients reported moderate to moderate-high levels at baseline of Physical Functioning, Role Functioning, and Global Health Status/Quality of Life (QoL) and low levels of fatigue (weakness, tiredness) as measured by the EORTC QLQ-C30 at baseline which were maintained during treatment. Most patients indicated that symptoms commonly associated with Columvi treatment (constipation, diarrhoea, and nausea) were not present or were of low severity if present, and maintained during treatment. Patients reported low levels of lymphoma symptoms at baseline as measured by the FACT-Lym scale which were maintained during treatment.
Columvi in combination with gemcitabine and oxaliplatin. The efficacy of Columvi in combination with gemcitabine and oxaliplatin (Columvi + GemOx) was evaluated in study GO41944 (STARGLO), an open-label multicentre, randomised clinical trial that included 274 patients with relapsed or refractory DLBCL not otherwise specified (DLBCL NOS). The study excluded patients who received only one prior line of therapy who were candidates for stem cell transplant, patients with HGBCL, PMBCL, history of transformation of indolent disease to DLBCL, prior allogeneic haematopoietic stem cell transplant, previous or active CNS lymphoma, ECOG performance status > 2, CrCl < 30 mL/min, or hepatic transaminases > 2.5 x ULN.
Patients were randomised 2:1 to receive Columvi + GemOx (N = 183) or rituximab in combination with gemcitabine and oxaliplatin (R-GemOx; N = 91) for 8 cycles, followed by 4 additional cycles of Columvi monotherapy for patients in the Columvi + GemOx arm. Randomisation was stratified by number of previous lines of systematic therapy for DLBCL (1 vs. ≥ 2) and outcome of last systematic therapy (relapsed vs. refractory).
In the Columvi + GemOx arm, following pre-treatment with obinutuzumab at cycle 1 day 1, patients received 2.5 mg of Columvi at cycle 1 day 8, 10 mg of Columvi at cycle 1 day 15, and 30 mg of Columvi at cycle 2 day 1 as per the step-up dosing schedule. Patients continued to receive 30 mg of Columvi on day 1 of cycles 3 to 12. Patients received premedication including an anti-pyretic, an anti-histamine, and dexamethasone. For patients who were unable to receive dexamethasone, an alternate corticosteroid was used (see Section 4.2 Dose and Method of Administration). Gemcitabine (1000 mg/m2) and oxaliplatin (100 mg/m2) were administered intravenously on day 2 of cycle 1 and then on day 1 of subsequent cycles, up to cycle 8. The duration of each cycle was 21 days. G-CSF primary prophylaxis was mandatory for the initial 2 cycles.
Patients received a median of 11 cycles of Columvi treatment (range: 1 to 13 cycles).
The baseline demographic and disease characteristics were: median age 68 years (range: 20 to 88 years); 57.7% males; 50.0% Asian, 42.0% white, 5.8% Hispanic or Latino and 1.1% Black or African American; and ECOG performance status of 0 (43.3%), 1 (46.6%), or 2 (10.1%). The majority of patients (62.8%) had received 1 prior line of systemic therapy; 37.2% of patients had received 2 or more prior lines. All patients had received prior chemotherapy and most (98.5%) had received anti-CD20 monoclonal antibody therapy; 7.7% of patients had received CAR T-cell therapy, and 4.0% had received autologous stem cell transplant. The majority of patients (66.8%) had refractory disease; 55.8% had primary refractory disease, and 60.6% of patients were refractory to their last prior therapy.
The primary efficacy outcome measure was overall survival (OS). At the time of the prespecified primary analysis, a statistically significant improvement in OS was observed for patients randomised to the Columvi + GemOx arm compared to patients randomised to R-GemOx. Statistically significant improvements in PFS and CR rate, as assessed by an Independent Review Committee (IRC), were also observed with Columvi + GemOx over R-GemOx.
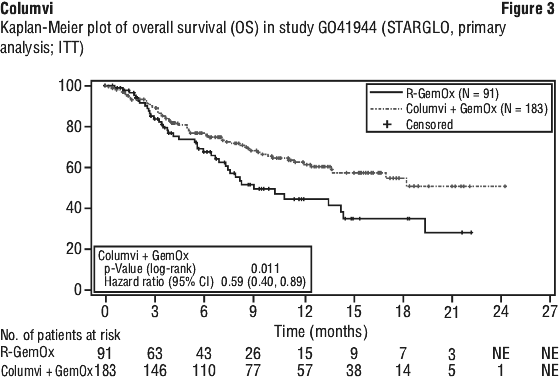
Overall survival, PFS, and CR results from an updated analysis conducted after an additional 10.5 months of follow-up continue to demonstrate benefit of Columvi + GemOx over R-GemOx. Median OS was 25.5 months in the Columvi + GemOx arm compared with 12.9 months in the R-GemOx arm in the updated analysis (HR of 0.62, 95% CI: 0.43, 0.88). The 24-month OS rate was 52.8% (95% CI: 44.8, 60.7) in the Columvi + GemOx arm and 33.5% (95% CI: 22.2, 44.9) in the R-GemOx arm. Median PFS was 13.8 months in the Columvi + GemOx arm compared with 3.6 months in the R-GemOx arm (HR of 0.40, 95% CI: 0.28, 0.57) and CRR (%) was 58.5 in the Columvi + GemOx arm compared with 25.3 in the R-GemOx arm (difference in response rate of 33.2%, 95% CI: 20.9, 45.5) in the updated analysis.
Pre-specified exploratory subgroup analyses raised a hypothesis of differential efficacy across geographical regions but were not powered to detect statistical significance. Results for Australian patients (n = 30) did not appear meaningfully different from those for the overall study population. See Figure 3.
Efficacy results are summarised in Table 11.
Immunogenicity.
As with all therapeutic proteins, there is a potential immunogenicity.
Across studies, the majority of patients (574/608 patients; 94.4%) who received Columvi were negative for antidrug antibodies (ADAs) at baseline and remained negative throughout treatment with Columvi. Four (0.7%) patients were negative for ADAs at baseline and became positive for ADAs during treatment. Four patients (0.7%) were ADA-positive at baseline and at one or more post-dose timepoints. Due to the limited number of patients with antibodies against Columvi, no conclusions can be drawn concerning a potential effect of immunogenicity on efficacy or safety.
The detection of an immune response is highly dependent on the sensitivity and specificity of the assays used, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Columvi with the incidence of antibodies to other products may be misleading.
5.2 Pharmacokinetic Properties
Non-compartmental analyses indicate that glofitamab serum concentration reaches the maximal level (Cmax) at the end of infusion and declines in a bi-exponential fashion. Glofitamab exhibits linear and dose-proportional pharmacokinetics over the dose range studied (0.005 to 30 mg) and is independent of time.
Absorption.
Columvi is administered as an IV infusion. Peak concentration of glofitamab (Cmax) was reached at the end of the infusion.
Distribution.
Following IV administration, the central volume of distribution was 3.34 L, which is close to total serum volume. The peripheral volume of distribution was 2.35 L.
Metabolism.
The metabolism of glofitamab has not been directly studied. Antibodies are cleared principally by catabolism.
Excretion.
The glofitamab serum concentration-time data are described by a population pharmacokinetic model with two compartments and both time-independent clearance and time-varying clearance.
The time-independent clearance pathway was estimated as 0.633 L/day and the initial time-varying clearance pathway as 0.814 L/day, with an exponential decay over time (Kdes ~ 1.5/day). The estimated decay half-life from the initial total clearance value to the time-independent clearance only was estimated as 0.471 days.
The effective half-life in the linear phase (i.e. after the contribution of time-varying clearance has collapsed to a negligible amount) is 7.92 days (geometric mean, 95% CI: 4.69, 11.90) based on the population pharmacokinetic analysis.
Pharmacokinetics in special populations.
Paediatric population.
No studies have been conducted to investigate the pharmacokinetics of glofitamab in paediatric patients.
Geriatric population.
No differences in glofitamab exposure were noted in patients 65 years of age and older and those under 65 years based on population pharmacokinetic analysis.
Renal impairment.
Population pharmacokinetic analyses showed that creatinine clearance does not affect the pharmacokinetics of glofitamab. The pharmacokinetics of glofitamab in patients with mild or moderate renal impairment (CrCl 30 to < 90 mL/min) were similar to those in patients with normal renal function. No dose adjustment is required for patients with mild or moderate renal impairment. Columvi has not been studied in patients with severe renal impairment.
Hepatic impairment.
Population pharmacokinetic analyses showed hepatic impairment does not affect the pharmacokinetics of glofitamab. The pharmacokinetic of glofitamab in patients with mild hepatic impairment (total bilirubin > ULN to ≤ 1.5 x ULN or AST > ULN) were similar to those with normal hepatic functions. No dose adjustment is required for patients with mild hepatic impairment. Columvi has not been studied in patients with moderate and severe hepatic impairment.
5.3 Preclinical Safety Data
Genotoxicity.
No genotoxicity studies have been performed with glofitamab. As a large protein molecule, glofitamab is not expected to interact with DNA or other chromosomal material.
Carcinogenicity.
No long-term animal studies have been performed to investigate the carcinogenic potential of glofitamab.6 Pharmaceutical Particulars
6.1 List of Excipients
Histidine, histidine hydrochloride monohydrate, methionine, sucrose, polysorbate 20, water for injections.
6.2 Incompatibilities
Only 0.9% or 0.45% sodium chloride solution should be used to dilute Columvi, since other diluents have not been tested.
Columvi when diluted with 0.9% sodium chloride solution, is compatible with intravenous infusion bags composed of polyvinyl chloride (PVC), polyethylene (PE), polypropylene (PP), or non-PVC polyolefin. When diluted with 0.45% sodium chloride solution, Columvi is compatible with intravenous infusion bags composed of PVC.
No incompatibilities have been observed with infusion sets with product-contacting surfaces of polyurethane (PUR), PVC, or PE, and in-line filter membranes composed of polyethersulfone (PES) or polysulfone. The use of in-line filter membranes is optional.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
Shelf life of diluted solution for intravenous infusion.
The prepared infusion solution should be used immediately. If not used immediately, the infusion solution can be stored in the refrigerator at 2°C to 8°C for up to 64 hours or at 25°C for up to 4 hours.
6.4 Special Precautions for Storage
Vials.
Store at 2°C to 8°C.
Keep vial in the outer carton in order to protect from light.
Do not freeze. Do not shake.
6.5 Nature and Contents of Container
2.5 mL concentrate in a 6 mL vial (colourless Type I glass) with stopper (butyl rubber).
Pack size of 1 vial.
10 mL concentrate in a 15 mL vial (colourless Type I glass) with stopper (butyl rubber).
Pack size of 1 vial.
6.6 Special Precautions for Disposal
Do not shake the vial.
Instructions for dilution.
Columvi is for single use in one patient only. Discard any residue.
Columvi must be diluted by a healthcare professional using aseptic technique, prior to intravenous administration.
Visually inspect the Columvi vial for particulate matter or discoloration prior to administration. Columvi is a colourless, clear solution. Discard the vial if the solution is cloudy, discoloured, or contains visible particles.
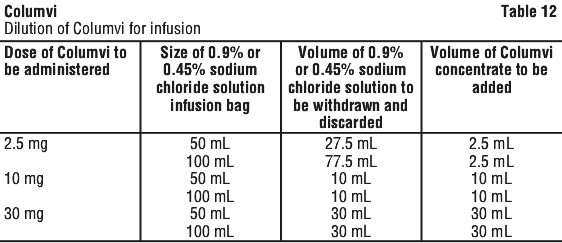
Withdraw the required volume of 0.9% or 0.45% sodium chloride solution from the infusion bag (see Table 12) using a sterile needle and syringe and discard.
Withdraw the required volume of Columvi concentrate for the intended dose from the vial using a sterile needle and syringe and dilute into the infusion bag (see Table 12). Discard any unused portion left in the vial.
The final drug concentration after dilution must be 0.1 mg/mL to 0.6 mg/mL.
Gently invert the infusion bag to mix the solution in order to avoid excessive foaming. Do not shake.
Inspect the infusion bag for particulates and discard if present.
Prior to the start of the intravenous infusion, the content of the infusion bag should be at room temperature.
Disposal.
The release of pharmaceuticals in the environment should be minimised. Medicines should not be disposed of via wastewater, and disposal through household waste should be avoided.6.7 Physicochemical Properties
Glofitamab is a humanised anti-CD20 anti-CD3 bispecific monoclonal antibody produced in Chinese hamster ovary (CHO) cells by recombinant DNA technology.
CAS number.
2229047-91-8.7 Medicine Schedule (Poisons Standard)
Prescription Only Medicine.
Summary Table of Changes
