Notes
Distributed by Arrotex Pharmaceuticals
1 Name of Medicine
Corora is the Amgen Inc. trademark for denosumab (rch).
2 Qualitative and Quantitative Composition
Each 1 mL single-use pre-filled syringe contains 60 mg denosumab.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Corora is a sterile, preservative-free, clear, colourless to slightly yellow solution for injection at pH 5.2. The solution may contain trace amounts of translucent to white proteinaceous particles.
4.1 Therapeutic Indications
The treatment of osteoporosis in postmenopausal women. Corora significantly reduces the risk of vertebral, non-vertebral and hip fractures.
Treatment to increase bone mass in men with osteopaenia receiving androgen deprivation therapy for non-metastatic prostate cancer (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Treatment to increase bone mass in men with osteoporosis at increased risk of fracture.
Treatment to increase bone mass in women and men at increased risk of fracture due to long-term systemic glucocorticoid therapy.
4.2 Dose and Method of Administration
Dosage (dose and interval).
Administration should be performed by an individual who has been adequately trained in injection techniques.
The recommended dose of Corora is a single subcutaneous (SC) injection of 60 mg, once every 6 months. If Corora treatment is discontinued, consideration should be given to transitioning to an alternative antiresorptive therapy.
To reduce the risk of hypocalcaemia, patients must be adequately supplemented with calcium and vitamin D (see Section 4.4 Special Warnings and Precautions for Use, Hypocalcaemia). In the major clinical trials of Corora, daily supplementation with 1,000 mg of calcium and at least 400 IU vitamin D was recommended.
Method of administration.
For subcutaneous use.
Corora is a sterile and preservative-free product. Before administration, the Corora solution should be inspected for particulate matter and discolouration. Do not use if the solution is cloudy or discoloured. Do not excessively shake the pre-filled syringe. To avoid discomfort at the site of injection, allow the pre-filled syringe to reach room temperature (up to 25°C) before injecting, and inject slowly. Inject the entire contents of the pre-filled syringe.
Product is for single-use in one patient only. Dispose of any medicinal product remaining in the pre-filled syringe.
Dosage adjustment.
Elderly patients.
No dose adjustment is necessary in elderly patients (see Section 4.4 Special Warnings and Precautions for Use, Use in the elderly).
Renal impairment.
No dose adjustment is necessary in patients with renal impairment (see Section 4.4 Special Warnings and Precautions for Use, Use in renal impairment).4.3 Contraindications
Hypocalcaemia (see Section 4.4 Special Warnings and Precautions for Use).
Hypersensitivity to the active substance, to CHO-derived proteins or to any of the excipients (see Section 6.1 List of Excipients).
Pregnancy and in women trying to get pregnant (see Section 4.6 Fertility, Pregnancy and Lactation).
4.4 Special Warnings and Precautions for Use
Hypocalcaemia.
Hypocalcaemia must be corrected prior to initiating therapy with Corora. In the post-marketing setting, severe symptomatic hypocalcaemia (resulting in hospitalisation, life-threatening events and fatal cases) has been reported (see Section 4.8 Adverse Effects (Undesirable Effects)), particularly in patients with severe renal impairment, receiving dialysis or treatment with other calcium lowering drugs. While most cases occurred in the first few weeks of initiating therapy, hypocalcemia has also occurred later. Clinical monitoring of calcium levels is recommended before each dose.
In patients predisposed to hypocalcaemia (e.g. history of hypoparathyroidism, thyroid surgery, parathyroid surgery, malabsorption syndromes, excision of small intestine, severe renal impairment [creatinine clearance CrCl < 30 mL/min], receiving dialysis or treatment with other calcium lowering drugs), clinical monitoring of calcium levels is recommended during treatment, especially in the first two weeks of initiating therapy.
Hypocalcaemia following Corora administration is a significant risk in patients with severe renal impairment [creatinine clearance CrCl < 30 mL/min], receiving dialysis or treatment with other calcium lowering drugs. These patients may also develop marked elevations of serum parathyroid hormone (PTH). Concomitant use of calcimimetic drugs may worsen the risk of hypocalcaemia.
Instruct all patients about the symptoms of hypocalcaemia and the importance of maintaining calcium levels with adequate calcium and vitamin D supplementation.
Adequate intake of calcium and vitamin D is important in all patients (see Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects)).
Skin infections.
Patients receiving Corora may develop skin infections (predominantly cellulitis) leading to hospitalisation (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be advised to seek prompt medical attention if they develop signs or symptoms of cellulitis.
Osteonecrosis of the jaw.
Osteonecrosis of the jaw (ONJ) has been reported in patients treated with denosumab or bisphosphonates, another class of antiresorptive agents. Most cases have been in cancer patients; however some have occurred in patients with osteoporosis.
ONJ has been reported rarely in clinical studies in patients receiving denosumab at a dose of 60 mg every 6 months for osteoporosis. There have been reports of ONJ in clinical studies in patients with advanced cancer treated with denosumab at the studied dose of 120 mg administered monthly.
Known risk factors for ONJ include a diagnosis of cancer with bone lesions, concomitant therapies (e.g. chemotherapy, antiangiogenic biologics, corticosteroids, radiotherapy to head and neck), poor oral hygiene, invasive dental procedures (e.g. tooth extraction), and co-morbid disorders (e.g. pre-existing dental disease, anaemia, coagulopathy, infection). The risk of ONJ may increase with duration of exposure to Corora.
It is important to evaluate patients for risk factors for ONJ before starting treatment. If risk factors are identified, a dental examination with appropriate preventive dentistry is recommended prior to treatment with Corora. Good oral hygiene practices should be maintained during treatment with Corora.
Avoid invasive dental procedures during treatment with Corora. For patients in whom invasive dental procedures cannot be avoided, the clinical judgement of the treating physician should guide the management plan of each patient based on individual benefit/risk assessment.
Patients who are suspected of having or who develop ONJ while on Corora should receive care by a dentist or an oral surgeon. In patients who develop ONJ during treatment with Corora, a temporary interruption of Corora treatment should be considered based on individual benefit/risk assessment until the condition resolves.
Atypical femoral fractures.
Atypical femoral fractures have been reported in patients receiving Corora. Atypical femoral fractures may occur with little or no trauma in the subtrochanteric and diaphyseal regions of the femur and may be bilateral. Specific radiographic findings characterise these events. Atypical femoral fractures have also been reported in patients with certain co-morbid conditions (e.g. vitamin D deficiency, rheumatoid arthritis, hypophosphatasia) and with use of certain pharmaceutical agents (e.g. bisphosphonates, glucocorticoids, proton pump inhibitors). These events have also occurred without antiresorptive therapy. During Corora treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Patients presenting with such symptoms should be evaluated for an incomplete femoral fracture, and the contralateral femur should also be examined.
Multiple vertebral fractures (MVF) following discontinuation of Corora treatment.
Multiple vertebral fractures (MVF) may occur following discontinuation of treatment with Corora, particularly in patients with a history of vertebral fracture. New vertebral fractures occurred as early as 7 months after the last dose of Corora.
Patients being treated with Corora, should be advised not to interrupt Corora therapy without prior consultation with their treating physician. The individual benefit/risk should be evaluated before discontinuing treatment with Corora. If Corora treatment is discontinued, consideration should be given to transitioning to an alternative antiresorptive therapy.
Hypercalcaemia in paediatric patients with osteogenesis imperfecta.
Corora is not indicated for use in paediatric patients. In clinical trials, hypercalcaemia has been reported very commonly in paediatric patients with osteogenesis imperfecta treated with denosumab. Some cases required hospitalisation and were complicated by acute renal injury (see Section 4.4 Special Warnings and Precautions for Use, Paediatric use).
Drugs with same active ingredient.
Corora contains the same active ingredient found in Xgeva (denosumab), used for the treatment of skeletal related events in patients with bone metastasis from solid tumours. Patients being treated with Corora should not be treated with Xgeva and/or other denosumab-containing medicines concomitantly.
Use in glucocorticoid induced osteoporosis.
In GIOP, fractures occur at a higher BMD than postmenopausal osteoporosis. There is limited data about the impact of denosumab on fractures in this setting.
Use in special populations.
Use in hepatic impairment.
The safety and efficacy of Corora has not been studied in patients with hepatic impairment.
Use in renal impairment.
No dose adjustment is necessary in patients with renal impairment.
In clinical studies, patients with severe renal impairment (creatinine clearance CrCl < 30 mL/min) or receiving dialysis were at greater risk of developing hypocalcaemia. Adequate intake of calcium and vitamin D is important in patients with severe renal impairment or receiving dialysis (see Section 4.4 Special Warnings and Precautions for Use, Hypocalcaemia).
Use in the elderly.
Of the total number of patients in clinical studies of Corora, 9,943 patients were ≥ 65 years, while 3,576 were ≥ 75 years. No overall differences in safety or efficacy were observed between these patients and younger patients.
Of the patients in the osteoporosis study in men, 133 patients (55%) were ≥ 65 years old, while 39 patients (16%) were ≥ 75 years old.
Paediatric use.
Corora is not indicated for use in paediatric patients. In clinical trials, hypercalcaemia has been reported very commonly in paediatric patients with osteogenesis imperfecta treated with denosumab. Some cases required hospitalisation and were complicated by acute renal injury (see Section 4.4 Special Warnings and Precautions for Use, Hypercalcaemia in paediatric patients with osteogenesis imperfecta).
Adolescent primates dosed with denosumab at 27 and 150 times (10 mg/kg and 50 mg/kg dose) the clinical exposure based on area under the curve (AUC) had abnormal growth plates. In neonatal rats, inhibition of RANKL (target of denosumab therapy) with a construct of osteoprotegerin bound to immunoglobulin Fc segment (OPG-Fc) at high doses was associated with inhibition of bone growth and tooth eruption. Therefore, treatment with denosumab may impair bone growth in children with open growth plates and may inhibit eruption of dentition.
Effects on laboratory tests.
No interactions with laboratory and diagnostic tests have been identified.4.5 Interactions with Other Medicines and Other Forms of Interactions
Calcimimetics.
Concomitant use of calcimimetic drugs (e.g. cinacalcet) may worsen the risk of hypocalcaemia.
In an interaction study conducted on 17 postmenopausal women with osteoporosis, midazolam (2 mg oral) was administered two weeks after a single dose of denosumab (60 mg subcutaneous injection), which approximates the Tmax of denosumab. Corora did not affect the pharmacokinetics of midazolam, which is metabolised by cytochrome P450 3A4 (CYP3A4). This indicates that Corora should not alter the pharmacokinetics of drug products metabolised by CYP3A4.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No data are available on the effect of denosumab on human fertility. Denosumab had no effect on female fertility or male reproductive organs or sperm motility in cynomolgus monkeys at subcutaneous doses up to 12.5 mg/kg/week (females) or 50 mg/kg/month (males), yielding exposures that were approximately 150-fold higher than the human exposure at 60 mg subcutaneous administered once every 6 months.
(Category D)
There are no adequate and well-controlled studies of Corora in pregnant women. Corora is contraindicated for use during pregnancy and in women trying to get pregnant. Premenopausal women with reproductive potential should be advised of the potential effects of Corora in pregnancy. Contraception should be discussed. Women should be advised not to become pregnant during and for at least 5 months after treatment with Corora.
Developmental toxicity studies have been performed with denosumab in cynomolgus monkeys and have shown serious adverse events on development (including foetal and infant lethality). Denosumab was shown to cross the placenta in monkeys (see Section 5.3 Preclinical Safety Data, Reproductive toxicity).
It is unknown whether denosumab is excreted in human milk. Only limited excretion of denosumab in milk was observed in a study in monkeys. A decision on whether to abstain from breast-feeding or to abstain from therapy with Corora should be made, taking into account the benefit of breast-feeding to the newborn/infant and the benefit of Corora therapy to the woman.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive or use machinery have been performed.
4.8 Adverse Effects (Undesirable Effects)
Treatment of postmenopausal osteoporosis.
Corora has been studied in over 10,500 women with postmenopausal osteoporosis in clinical trials representing up to 10 years of continued Corora treatment.
The safety of Corora in the treatment of postmenopausal osteoporosis was assessed in FREEDOM, a large, 3-year, randomised, double-blind, placebo-controlled, multinational phase III study of 7,808 postmenopausal women aged 60 to 91 years with osteoporosis. A total of 3,886 women were exposed to Corora and 3,876 women were exposed to placebo administered once every 6 months as a single 60 mg subcutaneous dose.
The safety of Corora was also assessed in a second phase 3 study of similar design. A total of 322 postmenopausal women aged 43 to 83 years with low bone mass were enrolled in this 2-year study. A total of 164 women were exposed to Corora and 165 women were exposed to placebo administered once every 6 months as a single 60 mg subcutaneous dose.
In both studies, all women received at least 1,000 mg of calcium and 400 IU of vitamin D supplementation per day.
Across the two phase III studies the incidence of all-cause mortality was 1.7% (n = 70) in the Corora group and 2.2% (n = 90) in the placebo group. The incidence of serious adverse events was 25.3% in the Corora group and 24.3% in the placebo group. The percentage of patients who withdrew from the studies due to adverse events was 2.3% and 2.1% for the Corora and placebo groups, respectively.
The most common adverse events reported in studies of women with postmenopausal osteoporosis or low bone mass (n = 8,091), occurring in ≥ 10% of patients either in the Corora-treated or placebo group, were back pain (34.1% Corora, 34.0% placebo), arthralgia (20.4% in each group), hypertension (15.3% Corora, 16.1% placebo), nasopharyngitis (14.8% Corora, 15.6% placebo), pain in extremity (11.8% Corora, 11.2% placebo) and osteoarthritis (10.9% Corora, 11.1% placebo).
Adverse events reported in at least 2% of postmenopausal women with osteoporosis or low bone mass (n = 8,091) and at least 1% more frequently in the Corora-treated women than in the placebo-treated women were: hypercholesterolaemia (7.0% Corora, 5.9% placebo) and eczema (includes dermatitis, allergic dermatitis, atopic dermatitis and contact dermatitis) (3.1% Corora, 1.7% placebo).
In STAND, a double-blind, randomised, alendronate-controlled, study in postmenopausal women with low bone mass who had received alendronate for at least 6 months preceding study entry, patients received either Corora 60 mg Q6M SC (n = 253) or alendronate orally 70 mg weekly for 12 months (n = 249). The safety profile was similar for patients transitioning from alendronate to denosumab and those continuing on alendronate therapy, including the overall incidence of adverse events and serious adverse events. Eight patients (3.2%) in the Corora group and 4 patients (1.6%) in the alendronate group reported adverse events of fracture.
Hypocalcaemia.
In two phase III placebo-controlled clinical trials in postmenopausal women with osteoporosis, approximately 0.05% (2 of 4,050) of patients had declines of serum calcium levels (less than 1.88 mmol/L) following Corora administration.
Skin infections.
In two phase III placebo-controlled clinical trials in postmenopausal women with osteoporosis, skin infections leading to hospitalisation were reported more frequently in the Corora (0.4%, 16 of 4,050) versus the placebo (0.1%, 3 of 4,041) groups, respectively. These cases were predominantly cellulitis. The overall incidence of skin infections was similar between the Corora (1.5%, 59 of 4,050) and placebo groups (1.2%, 50 of 4,041).
Pancreatitis.
Pancreatitis was reported in 4 patients (0.1%) in the placebo and 8 patients (0.2%) in the Corora groups. Several patients had a prior history of pancreatitis or a confounding event (e.g. gallstones). The time from product administration to event occurrence was variable.
Osteonecrosis of the jaw (ONJ).
In the osteoporosis clinical trial program, ONJ was reported rarely in patients treated with Corora.
Atypical femoral fractures.
In the osteoporosis clinical trial program, atypical femoral fractures were reported rarely in patients treated with Corora.
Multiple vertebral fractures (MVF) following discontinuation of Corora treatment.
In the osteoporosis clinical trial program, MVF were reported in patients following discontinuation of treatment with Corora, particularly in patients with a history of vertebral fracture.
Long-term safety in postmenopausal osteoporosis.
A total of 4,550 women who completed FREEDOM (Study 20030216, N = 7,808) enrolled into FREEDOM Extension (Study 20060289), a 7-year, multinational, multicentre, open-label, single-arm extension study to evaluate the long-term safety and efficacy of Corora. All patients in the extension study received Corora every 6 months as a single SC 60 mg dose, as well as daily calcium (1,000 mg) and vitamin D (at least 400 IU).
During the FREEDOM Extension study, the rates of adverse events observed through month 84 have not shown an increase over time and were similar to those observed in the initial 3 years of FREEDOM. Thirteen adjudicated cases of osteonecrosis of the jaw (ONJ) and two atypical fractures of the femur have occurred during the extension study.
Treatment of osteoporosis in men.
The safety of Corora in the treatment of men with osteoporosis was assessed in ADAMO, a randomised, double-blind, placebo-controlled study; a 1 year double-blind phase followed by a 1 year open-label extension. During the double-blind phase, a total of 120 men were exposed to Corora and 120 men were exposed to placebo administered subcutaneously once every 6 months as a single 60 mg dose. All men were instructed to take at least 1,000 mg of calcium and 800 IU of vitamin D supplementation per day.
The incidence of all-cause mortality was 0.8% (n = 1) in the Corora group and 0.8% (n = 1) in the placebo group. The incidence of serious adverse events was 9.2% in the Corora group and 8.3% in the placebo group. The percentage of patients who withdrew from the study due to adverse events was 2.5% and 0% for the Corora and placebo groups, respectively.
Adverse events in men with osteoporosis (n = 240) occurring in at least 5% of Corora-treated men and more frequently than in the placebo-treated patients were: back pain (8.3% Corora, 6.7% placebo), arthralgia (6.7% Corora, 5.8% placebo), and nasopharyngitis (6.7% Corora, 5.8% placebo).
Treatment of bone loss associated with androgen deprivation.
The safety of Corora in the treatment of bone loss associated with androgen deprivation in men with non-metastatic prostate cancer was assessed in a 3-year, randomised, double-blind, placebo-controlled, multinational study of 1,468 men aged 48 to 97 years. A total of 731 men were exposed to Corora and 725 men were exposed to placebo administered once every 6 months as a single 60 mg subcutaneous dose. The incidence of all-cause mortality was 6.0% (n = 44) in the Corora group and 6.3% (n = 46) in the placebo group. The incidence of serious adverse events was 34.6% in the Corora group and 30.6% in the placebo group. The percentage of patients who withdrew from the study due to adverse events was 7.0% and 6.1% for the Corora and placebo groups, respectively.
Adverse events reported in men with bone loss associated with androgen deprivation (n = 1,456) occurring in at least 2% of Corora-treated men and at least 1% more frequently in Corora-treated men than placebo-treated men were: arthralgia (12.6% Corora, 11.0% placebo), pain in extremity (9.0% Corora, 7.0% placebo), musculoskeletal pain (5.6% Corora, 3.6% placebo), dizziness (5.6% Corora, 4.3% placebo), metastases to bone (4.7% Corora, 3.4% placebo), osteoarthritis (4.2% Corora, 3.2% placebo), cataract (4.7% Corora, 1.2% placebo), bronchitis (4.1% Corora, 2.9% placebo), urinary retention (3.1% Corora, 1.5% placebo), angina pectoris (2.3% Corora, 1.1% placebo) and procedural pain (2.1% Corora, 0.4% placebo).
Treatment of bone loss associated with systemic glucocorticoid therapy.
The safety of Corora in the treatment of bone loss associated with systemic glucocorticoid therapy in men and women was assessed over the first 12 months of a 24 month, randomised, double-blind, double-dummy, active-controlled study. Subjects received either Corora 60 mg Q6M SC (n = 394) or risedronate orally 5 mg daily (n = 384). All subjects were instructed to take at least 1,000 mg of calcium and 800 IU of vitamin D supplementation per day.
The incidence of all-cause mortality during the first 12 months of the study was 1.5% (n = 6) in the Corora group and 0.5% (n = 2) in the risedronate group. Three additional deaths were reported for subjects in the risedronate group but were not included because it was not possible to confirm exposure to risedronate during the study. The incidence of serious adverse events was 16.0% in the Corora group and 16.9% in the risedronate group. The percentage of patients who withdrew from the study due to adverse events was 3.8% and 3.6% for the Corora and risedronate groups, respectively. The percentage of patients who discontinued investigational product due to adverse events was 6.3% and 7.6% for Corora and risedronate respectively.
Adverse events occurring in subjects during the first 12 months of the study in at least 3% of Corora-treated subjects and more frequently in the Corora-treated group were: back pain (4.6% Corora, 4.4% risedronate), hypertension (3.8% Corora, 3.4% risedronate), bronchitis (3.8% Corora, 2.9% risedronate), headache (3.6% Corora, 1.8% risedronate), dyspepsia (3.0% Corora, 2.6% risedronate), urinary tract infection (3.0% Corora, 2.1% risedronate), upper abdominal pain (3.0% Corora, 1.8% risedronate) and bone pain (1.0% Corora, 0% risedronate). Subject incidence of fractures are shown in Table 1.

Post-marketing experience.
Rare events of drug-related hypersensitivity reactions: rash, urticaria, facial swelling, erythema and anaphylactic reactions.
Rare events of severe symptomatic hypocalcaemia (resulting in hospitalisation, life-threatening events, and fatal cases) have been reported predominantly in patients at increased risk of hypocalcaemia, particularly in patients with severe renal impairment, receiving dialysis or treatment with other calcium lowering drugs receiving Corora. Most cases occurred in the first weeks of initiating therapy. Examples of the clinical manifestations of severe symptomatic hypocalcaemia have included QT prolongation, tetany, seizures and altered mental status (see Section 4.4 Special Warnings and Precautions for Use, Hypocalcaemia). Symptoms of hypocalcaemia in denosumab clinical studies included paraesthesia, muscle stiffness, twitching, spasms and muscle cramps.
Musculoskeletal pain, including severe cases, has been reported in patients receiving Corora. There have been reports of osteonecrosis of the external auditory canal in patients using denosumab.
Very rare events of hypersensitivity vasculitis.
Uncommon events of injection site reactions (including injection site pain) have been observed.
Uncommon events of lichenoid drug eruptions (e.g. lichen planus-like reactions) have been observed.
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome has been observed.
Common events of alopecia have been reported.
The development or progression of lens opacification events (cataracts) were comparable between patients treated with Corora and those receiving placebo for up to 12 months in a clinical study in men with non-metastatic prostate cancer with bone loss due to androgen deprivation therapy.
Reporting of suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.4.9 Overdose
There is no experience with overdosage with Corora. Corora has been administered in clinical studies using doses up to 180 mg every 4 weeks (cumulative doses up to 1,080 mg over 6 months), and no additional adverse effects were observed.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
Denosumab is a fully human IgG2 monoclonal antibody with high affinity and specificity for RANK ligand (RANKL).
5.1 Pharmacodynamic Properties
Mechanism of action.
RANKL exists as a transmembrane or soluble protein. RANKL is essential for the formation, function and survival of osteoclasts, the sole cell type responsible for bone resorption. Osteoclasts play an important role in bone loss associated with postmenopausal osteoporosis and hormone ablation. Denosumab binds with high affinity and specificity to RANKL, preventing RANKL from activating its only receptor, RANK, on the surface of osteoclasts and their precursors, independent of bone surface. Prevention of RANKL/RANK interaction inhibits osteoclast formation, function and survival, thereby decreasing bone resorption and increasing bone mass and strength in both cortical and trabecular bone.
Pharmacodynamics.
In clinical studies, treatment with 60 mg of Corora resulted in rapid reduction in the bone resorption marker serum type 1 C-telopeptides (CTX) within 6 hours of SC administration by approximately 70%, with reductions of approximately 85% occurring by 3 days. CTX reductions were maintained over the 6-month dosing interval. At the end of each dosing interval, CTX reductions were partially attenuated from maximal reduction of ≥ 87% to ≥ 45% (range 45% to 80%), reflecting the reversibility of the effects of Corora on bone remodelling once serum denosumab levels diminish. These effects were sustained with continued treatment. Consistent with the physiological coupling of bone formation and resorption in skeletal remodelling, subsequent reductions in bone formation markers (e.g. bone specific alkaline phosphatase [BSAP] and serum N-terminal propeptide of type 1 collagen [P1NP]) were observed beginning 1 month after the first dose of Corora.
Bone turnover markers (bone resorption and formation markers) generally reached pretreatment levels within 9 months after the last 60 mg subcutaneous dose. Upon re-initiation, the degree of inhibition of CTX by Corora was similar to those observed in patients initiating Corora.
In a clinical study of postmenopausal women with low bone mass (n = 504) who were previously treated with alendronate for a median of 3 years, those transitioning to receive Corora experienced additional reductions in serum CTX, compared with women who remained on alendronate. In this study, the changes in serum calcium were similar between the two groups.
Clinical trials.
Treatment of osteoporosis in postmenopausal women. Independent risk factors, for example, low bone mineral density (BMD), age, the existence of previous fracture, family history of hip fractures, high bone turnover and low body mass index (BMI) should be considered in order to identify women at increased risk of osteoporotic fractures who could benefit from treatment.
Fracture reduction evaluation of denosumab in osteoporosis every 6 months (FREEDOM).
The efficacy and safety of Corora in the treatment of postmenopausal osteoporosis was demonstrated in FREEDOM (Study 20030216), a 3-year, randomised, double-blind, placebo-controlled, multinational study of women with baseline BMD T-scores at the lumbar spine or total hip between -2.5 and -4.0. 7,808 women aged 60 to 91 years were enrolled of whom 23.6% had prevalent vertebral fractures. Women with other diseases or on therapies that may affect bone (e.g. rheumatoid arthritis, osteogenesis imperfecta and Paget's disease) were excluded from this study.
BMD and other individual risk factors were collected for women enrolled in the FREEDOM study. The mean absolute 10-year fracture probability for women enrolled was 18.60% (deciles: 7.9 - 32.4%) for major osteoporotic fracture and 7.22% (deciles: 1.4 - 14.9%) for hip fracture, as derived from FRAX, the WHO Fracture Risk Assessment Tool algorithm.
Women were randomised to receive subcutaneous injections of either Corora 60 mg (n = 3,902) or placebo (n = 3,906) once every 6 months. Women received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily. The primary efficacy variable was the incidence of new vertebral fractures. Secondary efficacy variables included the incidence of non-vertebral fractures and hip fractures, assessed at 3 years.
Effect on vertebral fractures.
Corora significantly reduced the risk of new vertebral fractures at 1, 2 and 3 years (p < 0.0001) (see Table 2).
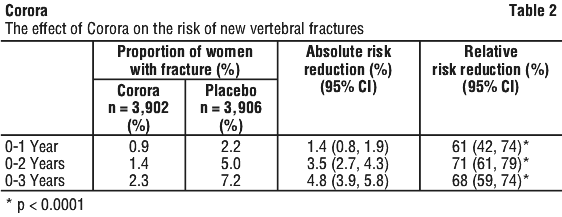 The reductions in the risk of new vertebral fractures by Corora over 3 years were consistent and significant regardless of whether or not women had a prevalent vertebral fracture or history of a non-vertebral fracture, and regardless of baseline age, BMD, bone turnover level and prior use of a medicinal product for osteoporosis.
The reductions in the risk of new vertebral fractures by Corora over 3 years were consistent and significant regardless of whether or not women had a prevalent vertebral fracture or history of a non-vertebral fracture, and regardless of baseline age, BMD, bone turnover level and prior use of a medicinal product for osteoporosis.
Corora also reduced the risk of new vertebral fracture by 65% (6.5% absolute risk reduction, p < 0.0001) in patients at high risk of fractures (defined as women who met ≥ 2 of the 3 following criteria at baseline: age ≥ 70 years, BMD T-score ≤ -3.0 [at lumbar spine, total hip, or femoral neck] or prevalent vertebral fracture).
Corora also reduced the risk of new and worsening vertebral fractures (67% relative risk reduction, 4.8% absolute risk reduction) as well as multiple vertebral fractures (61% relative risk reduction, 1.0% absolute risk reduction) at 3 years, when compared to placebo (all p < 0.0001).
Effect on hip fractures.
Corora demonstrated a 40% relative reduction (0.5% absolute risk reduction) in the risk of hip fracture over 3 years (p < 0.05) (see Figure 1). The incidence of hip fracture was 0.7% in the Corora group compared to 1.2% in the placebo group at 3 years.
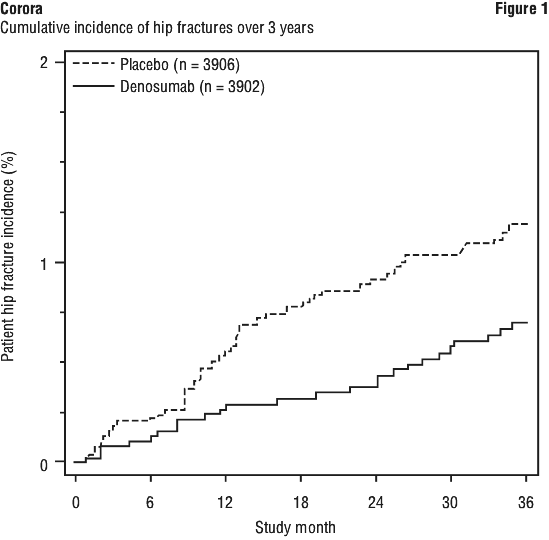 In women with high fracture risk as defined above by baseline age, BMD and prevalent vertebral fracture, a 48% relative risk reduction was observed with Corora (1.1% absolute risk reduction, p < 0.05).
In women with high fracture risk as defined above by baseline age, BMD and prevalent vertebral fracture, a 48% relative risk reduction was observed with Corora (1.1% absolute risk reduction, p < 0.05).
Effect on all clinical fractures.
Corora demonstrated superiority to placebo in reducing the risk of any clinical fractures, clinical (symptomatic) vertebral fractures, non-vertebral fractures (including hip), major non-vertebral fractures and major osteoporotic fractures (see Table 3).
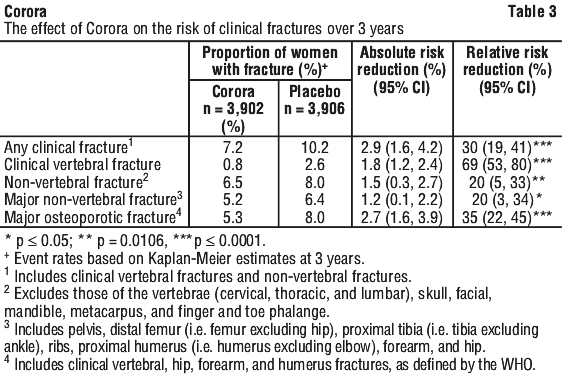 Women in the FREEDOM study had a mean baseline BMD T-score of -2.2 at the femoral neck. In women with baseline femoral neck BMD ≤ -2.5, Corora reduced the incidence of non-vertebral fracture (35% relative risk reduction, 4.1% absolute risk reduction, p < 0.001).
Women in the FREEDOM study had a mean baseline BMD T-score of -2.2 at the femoral neck. In women with baseline femoral neck BMD ≤ -2.5, Corora reduced the incidence of non-vertebral fracture (35% relative risk reduction, 4.1% absolute risk reduction, p < 0.001).
The reduction in the incidence of new vertebral fractures, hip fractures and non-vertebral fractures by Corora over 3 years were consistent regardless of the 10-year baseline fracture risk as assessed by FRAX.
Effect on bone mineral density.
Corora significantly increased BMD at all clinical sites measured, relative to treatment with placebo at 1, 2 and 3 years in FREEDOM. Corora increased BMD by 9.2% at the lumbar spine, 6.0% at the total hip, 4.8% at the femoral neck, 7.9% at the hip trochanter, 3.5% at the distal 1/3 radius and 4.1% at the total body over 3 years (all p < 0.0001). Increases in BMD at lumbar spine, total hip and hip trochanter were observed as early as 1 month after the initial dose. Corora increased lumbar spine BMD from baseline in 95% of postmenopausal women at 3 years. Consistent effects on BMD were observed at the lumbar spine regardless of baseline age, race, weight/BMI, BMD and bone turnover level. The effects of Corora on bone architecture were evaluated using quantitative computed tomography (QCT) in postmenopausal women with BMD T-score below -2.5 at the lumbar spine or total hip. Treatment with Corora increased volumetric trabecular BMD at the lumbar spine, volumetric BMD at the total hip and the volumetric cortical BMD and cortical thickness at the distal radius.
Study of Transitioning from Alendronate to Denosumab (STAND, Study 20050234) was a double-blind, randomised, alendronate-controlled, study in postmenopausal women with low BMD (T score between -2.0 and -4.0 at the lumbar spine or total hip) who had received alendronate (70 mg weekly [or equivalent] orally) for at least 6 months preceding study entry. Patients received either Corora 60 mg Q6M SC (n = 253) or alendronate orally 70 mg weekly for 12 months (n = 251).
Women who transitioned to receive Corora had greater increases in BMD at the total hip (1.9% versus 1.1%, p < 0.001; primary efficacy endpoint) after 1 year, compared to those who continued to receive alendronate therapy. Consistently greater increases in BMD were also seen at the lumbar spine, femoral neck, hip trochanter, and distal 1/3 radius in women treated with Corora, compared to those who continued to receive alendronate therapy (all p < 0.05).
In clinical studies examining the effects of discontinuation of Corora, BMD returned to approximately pre-treatment levels and remained above placebo within 18 months of the last dose. These data indicate that continued treatment with Corora is required to maintain the effect of the drug. Re-initiation of Corora resulted in gains in BMD similar to those when Corora was first administered.
Open-label extension study in the treatment of postmenopausal osteoporosis (FREEDOM extension study).
A total of 4,550 women, (2,343 Corora and 2,207 placebo) who missed no more than one dose of Corora in the FREEDOM pivotal study (Study 20030216, N = 7,808) and completed the month 36 study visit, enrolled in FREEDOM Extension (Study 20060289), a 7-year, multinational, multicentre, open-label, single-arm extension study to evaluate the long-term safety and efficacy of Corora. All women in the FREEDOM Extension study were to receive Corora every 6 months in an open-label manner as a single 60 mg SC dose, as well as daily calcium (at least 1,000 mg) and vitamin D (at least 400 IU). Safety was the primary endpoint; BMD and fracture incidence were two of the many secondary endpoints. At month 84 of the extension study, after 10 years of Corora treatment, the long-term group increased BMD by 21.7% (95% CI: 21.2, 22.2) at the lumbar spine, 9.2% (8.9, 9.5) at the total hip, 9.0% (8.6, 9.4) at the femoral neck and 13.0% (12.6, 13.4) at the trochanter from the pivotal FREEDOM study baseline. In years 4 through 10 of Corora treatment, the rates of new vertebral and non-vertebral fractures did not increase over time; annualised rates were approximately 1.0% and 1.3% respectively.
Bone histology.
Fifty-three transiliac crest bone biopsy specimens were obtained at either 2 years and/or 3 years from 47 postmenopausal women with osteoporosis treated with Corora in the FREEDOM study. Fifteen bone biopsy specimens were also obtained after 1 year of treatment with Corora from 15 postmenopausal women with low bone mass who had transitioned from previous alendronate therapy. Histology assessments in both studies showed bone of normal architecture and quality, as well as the expected decrease in bone turnover relative to placebo treatment. There was no evidence of mineralisation defects, woven bone or marrow fibrosis.
Fifty-nine women participated in the bone biopsy sub-study at month 24 (N = 41) and/or month 84 (N = 22) of the FREEDOM extension study, representing up to 5 and 10 years of treatment with Corora, respectively. Bone biopsy results showed bone of normal architecture and quality with no evidence of mineralisation defects, woven bone or marrow fibrosis as well as the expected decrease in bone turnover.
Histomorphometry findings in the FREEDOM extension study in postmenopausal women with osteoporosis showed that the antiresorptive effects of Corora, as measured by activation frequency and bone formation rates, were maintained over time.
Treatment of osteoporosis in men. A multicentre randomised double-blind placebo-controlled study to compare the efficacy and safety of denosumab versus placebo in males with osteoporosis (ADAMO).
The efficacy and safety of Corora in the treatment of men with osteoporosis was demonstrated in ADAMO (Study 20080098), a 1-year, multinational study of men with low bone mass, who had a baseline BMD T-score between -2.0 and -3.5 at the lumbar spine or femoral neck. Men with a BMD T-score between -1.0 and -3.5 at the lumbar spine or femoral neck and with history of prior fragility fracture were also enrolled. Men with other diseases (such as rheumatoid arthritis, osteogenesis imperfecta, and Paget's disease), or with significantly impaired renal function (GFR of ≤ 30 mL/min), or on therapies that may affect bone were excluded from this study. See Table 4.
 The 242 men enrolled in the ADAMO study ranged in age from 31 to 84 years and were randomised to receive subcutaneous injections of either Corora 60 mg (n = 121) or placebo (n = 121) once every 6 months. Men received calcium (at least 1,000 mg) and vitamin D (at least 800 IU) supplementation daily.
The 242 men enrolled in the ADAMO study ranged in age from 31 to 84 years and were randomised to receive subcutaneous injections of either Corora 60 mg (n = 121) or placebo (n = 121) once every 6 months. Men received calcium (at least 1,000 mg) and vitamin D (at least 800 IU) supplementation daily.
The primary efficacy variable was percent change in lumbar spine BMD at 1 year. Secondary efficacy variables included percent change in total hip, hip trochanter, femoral neck, and distal 1/3 radius BMD at 1 year, and change in CTX at day 15.
Corora significantly increased BMD at all clinical sites measured, relative to treatment with placebo at 1 year in men with osteoporosis. Corora increased BMD by 4.8% at the lumbar spine, 2.0% at the total hip, 2.3% at the hip trochanter, 2.2% at the femoral neck and 0.9% at the distal 1/3 radius, relative to placebo. Consistent effects on BMD were observed at the lumbar spine regardless of baseline age, race, weight/body mass index (BMI), BMD, and level of bone turnover.
Bone histology.
A total of 29 trans-iliac crest bone biopsy specimens were obtained from men with osteoporosis at 12 months (17 specimens in Corora group, 12 specimens in placebo group). Qualitative histology assessments showed normal architecture and quality with no evidence of mineralisation defects, woven bone, or marrow fibrosis.
Treatment of bone loss associated with androgen deprivation. The efficacy and safety of Corora in the treatment of bone loss associated with androgen deprivation was assessed in a 3-year randomised, double-blind, placebo-controlled, multinational study of 1,468 men with non-metastatic prostate cancer aged 48 to 97 years. All men regardless of age had histologically confirmed prostate cancer. Men less than 70 years of age also had either a BMD T-score at the lumbar spine, total hip, or femoral neck < -1.0 or a history of an osteoporotic fracture. Men over the age of 70 years did not have to meet the latter requirements. Men were randomised to receive subcutaneous injections of either Corora 60 mg (n = 734) or placebo (n = 734) once every 6 months. All men received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily. The primary efficacy variable was percent change in lumbar spine BMD.
Independent risk factors for osteoporosis other than BMD and advanced age (> 70 years of age) in males undergoing androgen deprivation, such as family history of hip fracture, alcohol or tobacco use, have not been validated to the same extent as females with postmenopausal osteoporosis. See Tables 5, 6 and 7.
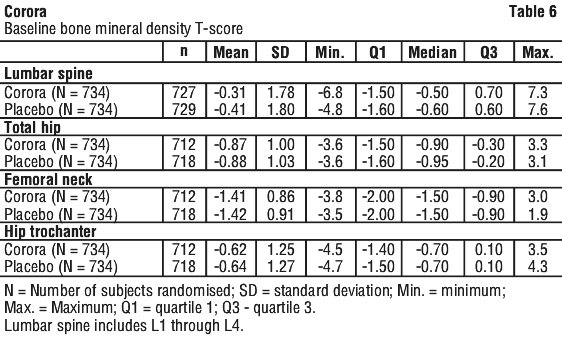
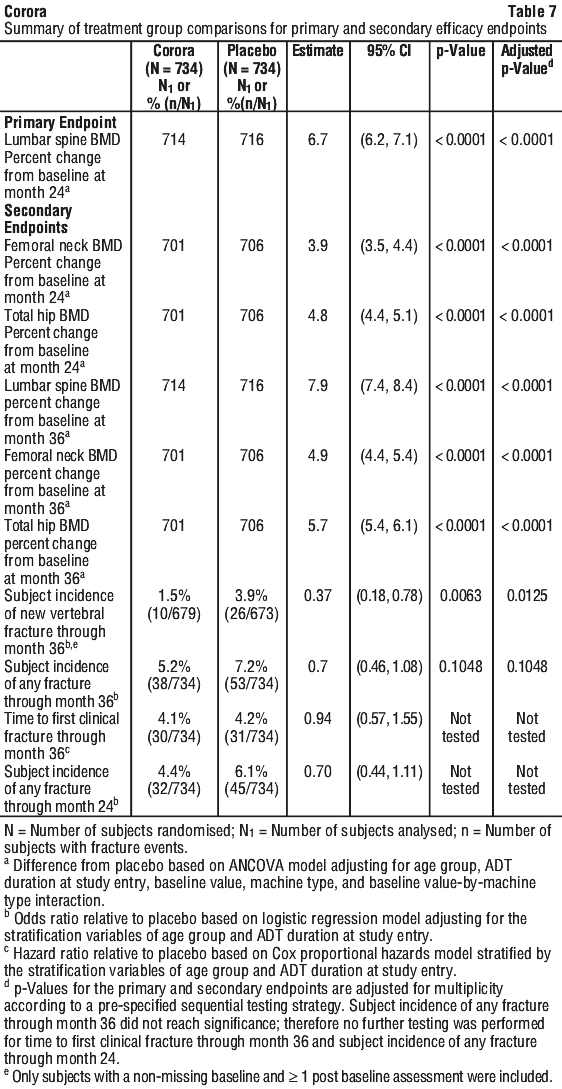 Corora significantly increased BMD at all clinical sites measured, relative to treatment with placebo at 3 years: 7.9% at the lumbar spine, 5.7% at the total hip, 4.9% at the femoral neck, 6.9% at the hip trochanter, 6.9% at the distal 1/3 radius and 4.7% at the total body (all p < 0.0001). Significant increases in BMD were observed at the lumbar spine, total hip, femoral neck and the hip trochanter as early as 1 month after the initial dose. Consistent effects on BMD were observed at the lumbar spine across subgroups of men regardless of baseline age, race, geographical region, weight/BMI, lumbar spine BMD T-score, bone turnover level; duration of androgen deprivation and presence of vertebral fracture at baseline.
Corora significantly increased BMD at all clinical sites measured, relative to treatment with placebo at 3 years: 7.9% at the lumbar spine, 5.7% at the total hip, 4.9% at the femoral neck, 6.9% at the hip trochanter, 6.9% at the distal 1/3 radius and 4.7% at the total body (all p < 0.0001). Significant increases in BMD were observed at the lumbar spine, total hip, femoral neck and the hip trochanter as early as 1 month after the initial dose. Consistent effects on BMD were observed at the lumbar spine across subgroups of men regardless of baseline age, race, geographical region, weight/BMI, lumbar spine BMD T-score, bone turnover level; duration of androgen deprivation and presence of vertebral fracture at baseline.
Corora demonstrated a significant relative risk reduction of new vertebral fractures as early as 1 year: 85% (1.6% absolute risk reduction) at 1 year, 69% (2.2% absolute risk reduction) at 2 years and 62% (2.4% absolute risk reduction) at 3 years (all p < 0.01).
Treatment of bone loss associated with systemic glucocorticoid therapy. The efficacy and safety of Corora in the treatment of bone loss associated with systemic glucocorticoid therapy were demonstrated by the 12-month primary analysis of a 24 month randomised, multicentre, double-blind, double-dummy, parallel-group, active-controlled study of 795 patients (70% women and 30% men) aged 20 to 94 years (mean age of 63.1 years) treated with ≥ 7.5 mg daily oral prednisone (or equivalent). The primary efficacy objective of the study was to demonstrate non-inferiority of Corora to oral risedronate with respect to percentage change from baseline in lumbar spine BMD at 12 months. The secondary objectives were to compare percentage change from baseline in lumbar spine and total hip BMD between Corora and risedronate at 12 and 24 months.
Two subpopulations were studied: glucocorticoid-continuing (≥ 7.5 mg daily prednisone or its equivalent for ≥ 3 months prior to study enrollment and planning to continue treatment for a total of at least 6 months; n = 505) and glucocorticoid-initiating (≥ 7.5 mg daily prednisone or its equivalent for < 3 months prior to study enrollment and planning to continue treatment for a total of at least 6 months; n = 290). Within each subpopulation, randomisation was stratified by gender and patients were randomised (1:1) to receive either Corora 60 mg subcutaneously once every 6 months (n = 398) or oral risedronate 5 mg once daily (active control) (n = 397). All patients were to receive at least 1,000 mg calcium and 800 IU vitamin D supplementation daily.
Enrolled patients < 50 years of age were required to have a history of osteoporotic fracture. Enrolled patients ≥ 50 years of age who were in the glucocorticoid-continuing subpopulation were required to have a baseline BMD T-score of ≤ -2.0 at the lumbar spine, total hip, or femoral neck; or a BMD T-score ≤ -1.0 at the lumbar spine, total hip, or femoral neck and a history of osteoporotic fracture. See Table 8.
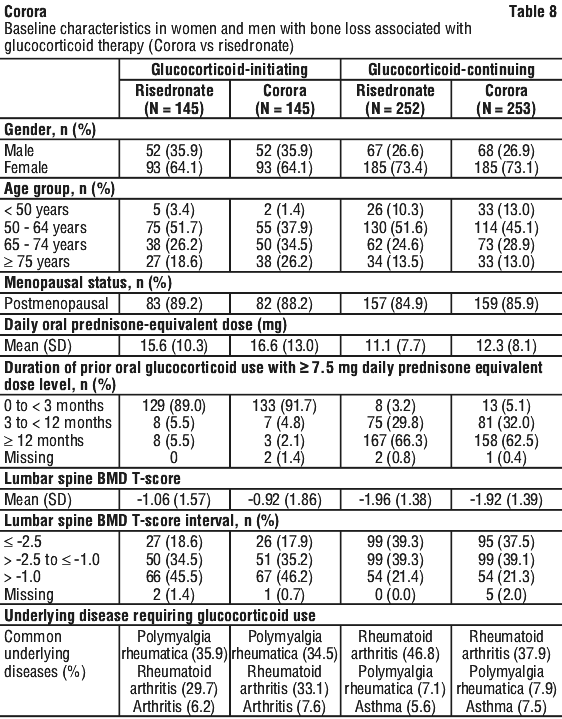
Effect on bone mineral density (BMD).
The difference in mean percentage change from baseline in lumbar spine BMD at 12 months between treatment groups (Corora - risedronate) was 2.2% (95% CI: 1.4, 3.0) in the glucocorticoid-continuing subpopulation and 2.9% (95% CI: 2.0, 3.9) in the glucocorticoid-initiating subpopulation, confirming non-inferiority.
The percentage change from baseline in lumbar spine and total hip BMD at 12 months was significantly greater with denosumab treatment than with risedronate treatment in both subpopulations (p < 0.001 in all comparisons) (see Table 9).
Consistent effects on lumbar spine BMD were observed regardless of gender; race; geographic region; menopausal status; age; and baseline lumbar spine BMD T-score, and glucocorticoid dose within each subpopulation.
In addition, exploratory endpoints measured the percentage change from baseline in femoral neck, hip trochanter and distal 1/3 radius BMD at 12 months (see Table 9). The study was not powered for reduction in risk of fracture. The correlation between increased bone mineral density and reduction of bone fracture incidence in patients with glucocorticoid-induced osteoporosis has not been directly established.
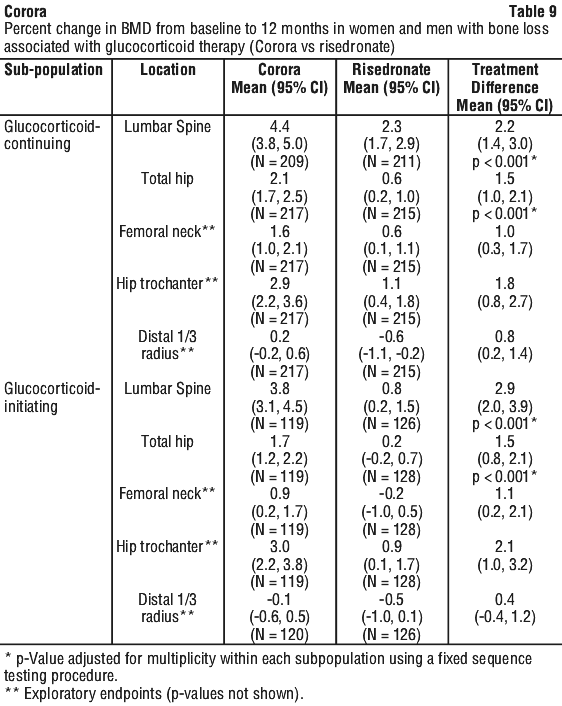
Bone histology.
Bone biopsy specimens evaluable for histology were obtained from 17 patients (6 in the Corora treatment group and 11 in the risedronate treatment group) at month 12. Qualitative histology assessments showed normal architecture and quality with no evidence of mineralisation defects, woven bone, or marrow fibrosis in patients treated with Corora.
5.2 Pharmacokinetic Properties
Absorption.
Following a 60 mg subcutaneous dose of denosumab, bioavailability was 61% and maximum serum denosumab concentrations (Cmax) of 6 microgram/mL (range 1 - 17 microgram/mL) occurred in 10 days (range 2 - 28 days).
Metabolism.
Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin and is unlikely to be eliminated via hepatic metabolic mechanisms. Based on nonclinical data, its metabolism and elimination are expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Excretion.
After Cmax, serum levels declined with a half-life of 26 days (range 6 - 52 days) over a period of 3 months (range 1.5 - 4.5 months). Fifty-three percent of patients had no measurable amounts of denosumab detected at 6 months post-dose.
No accumulation or change in denosumab pharmacokinetics over time was observed upon subcutaneous multiple-dosing of 60 mg once every 6 months. Denosumab pharmacokinetics was not affected by the formation of binding antibodies to denosumab and was similar in men and women.
Pharmacokinetic analysis was performed to evaluate the effects of demographic characteristics. This analysis showed no notable difference in pharmacokinetics with age (28 to 87 years), race or body weight (36 to 140 kg), or disease state (low bone mass or osteoporosis; prostate cancer).
Special populations.
Elderly.
The pharmacokinetics of denosumab was not affected by age (28 to 87 years).
Paediatric.
The pharmacokinetic profile has not been assessed in those ≤ 18 years.
Impaired hepatic function.
The pharmacokinetic profile has not been assessed in patients with impaired hepatic function.
Impaired renal function.
In a study of 55 patients with varying degrees of renal function, including patients on dialysis, the degree of renal impairment had no effect on the pharmacokinetics of denosumab (see Section 4.4 Special Warnings and Precautions for Use, Hypocalcaemia, and Use in renal impairment).
Immunogenicity.
In clinical studies, no neutralising antibodies for denosumab have been observed. Using a sensitive immunoassay < 1% of patients treated with denosumab for up to 5 years tested positive for non neutralising binding antibodies with no evidence of altered pharmacokinetics, toxicity, or clinical response.
5.3 Preclinical Safety Data
Genotoxicity.
The genotoxic potential of denosumab has not been evaluated. Denosumab is a recombinant protein comprised entirely of naturally occurring amino acids and contains no inorganic or synthetic organic linkers or other non-protein portions. Therefore, it is unlikely that denosumab or any of its derived fragments would react with DNA or other chromosomal material.
Carcinogenicity.
The carcinogenic potential of denosumab has not been evaluated in long-term animal studies. In view of the mechanism of action of denosumab, it is unlikely that the molecule would be capable of inducing tumour development or proliferation.
Reproductive toxicity.
In a study of cynomolgus monkeys with denosumab at subcutaneous doses up to 12.5 mg/kg/week given during the period equivalent to the first trimester, and yielding AUC exposure up to 99-fold higher than the human dose (60 mg every 6 months), there was no evidence of maternal or foetal harm. In this study, foetal lymph nodes were not examined.
In another study of cynomolgus monkeys dosed with denosumab throughout pregnancy at 50 mg/kg/month, yielding AUC exposure 119-fold higher than the human exposure, there were increased stillbirths and postnatal mortality; abnormal bone growth resulting in reduced bone strength, almost complete obliteration of bone marrow spaces (leading to reduced haematopoiesis), and tooth malalignment, dental dysplasia and a shortened/straighter dental arch (although no effect on the pattern or date of tooth eruption); altered appearance of eyes (increased apparent size, exophthalmos); absence of peripheral lymph nodes; and decreased neonatal growth. Following a 6 month period after birth, bone-related changes showed incomplete recovery. The effects on lymph nodes, tooth malalignment and dental dysplasia persisted, and minimal to moderate mineralisation in multiple tissues was seen in one animal. There was no evidence of maternal harm prior to labour; adverse maternal effects occurred infrequently during labour. Maternal mammary gland development was normal. A no observed adverse effect level has not been established in animal studies and the findings are attributable to the primary pharmacological activity of denosumab.
Preclinical studies in RANK/RANKL knockout mice suggest absence of RANKL could interfere with the development of lymph nodes in the foetus. Knockout mice lacking RANK or RANKL also exhibited decreased body weight, reduced bone growth and a lack of tooth eruption. Similar phenotypic changes (inhibition of bone growth and tooth eruption) were observed in a study in neonatal rats using a surrogate for denosumab, the RANKL inhibitor osteoprotegerin bound to Fc (OPG-Fc). Therefore, treatment with denosumab may impair bone growth in children with open growth plates and may inhibit eruption of dentition. A study on the reversibility of the effects of OPG-Fc showed persistence or only partial recovery (assessed after 10 weeks).
Preclinical studies in RANK/RANKL knockout mice suggest absence of RANKL during pregnancy may interfere with maturation of the mammary gland leading to impaired lactation post-partum.6 Pharmaceutical Particulars
6.1 List of Excipients
Each 1 mL single-use pre-filled syringe contains: 47 mg sorbitol, 1 mg acetate, 0.1 mg polysorbate 20, sodium hydroxide for adjusting to pH 5.2, in Water for Injection, (USP).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
It is recommended to store pre-filled syringes in a refrigerator at 2°C to 8°C in the original carton. Do not freeze. Protect from direct light. Do not excessively shake the pre-filled syringe. Do not expose to temperatures above 25°C.
If removed from the refrigerator, Corora should be kept at room temperature (up to 25°C) in the original container and must be used within 30 days.
6.5 Nature and Contents of Container
Pre-filled syringe with automatic needle guard.
Pack size of one Type 1 glass syringe, presented in blistered packaging.
The pre-filled syringe with automatic needle guard is not made with natural rubber latex.
Pre-filled syringe*.
Pack size of one Type 1 glass syringe, presented in blistered or unblistered packaging.
The pre-filled syringe is not made with natural rubber latex.
* Not available in Australia.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
 Denosumab has an approximate molecular weight of 147 kDa and is produced in genetically engineered mammalian cells (Chinese Hamster Ovary, CHO cells).
Denosumab has an approximate molecular weight of 147 kDa and is produced in genetically engineered mammalian cells (Chinese Hamster Ovary, CHO cells).
CAS number.
615258-40-7.7 Medicine Schedule (Poisons Standard)
S4 Prescription Medicine.
Summary Table of Changes
