What is in this leaflet
This leaflet answers some common questions about COSENTYX.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine. You can also download the most up to date leaflet from www.novartis.com.au. Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking COSENTYX against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What COSENTYX is used for
COSENTYX is used for the treatment of the following inflammatory diseases:
- Plaque psoriasis
- Psoriatic arthritis
- Axial spondyloarthritis, including ankylosing spondylitis (axial spondyloarthritis with radiographic damage) and non-radiographic axial spondyloarthritis (axial spondyloarthritis without radiographic damage)
Plaque psoriasis
COSENTYX is used to treat a skin condition called 'plaque psoriasis' in adults.
Plaque psoriasis causes inflammation of the skin.
COSENTYX is used in adults with moderate to severe plaque psoriasis.
Psoriatic arthritis
COSENTYX is used to treat a condition called 'psoriatic arthritis'. The condition is an inflammatory disease of the joints, often accompanied by psoriasis.
Axial spondyloarthritis, including ankylosing spondylitis (axial spondyloarthritis with radiographic damage) and non-radiographic axial spondyloarthritis (axial spondyloarthritis without radiographic damage)
COSENTYX is used to treat a condition called 'ankylosing spondylitis' and 'non-radiographic axial spondyloarthritis' in adults. These conditions are inflammatory diseases primarily affecting the spine which causes inflammation of the spinal joints.
In patients with plaque psoriasis, psoriatic arthritis, and axial spondyloarthritis the body produces increased amounts of a protein called IL-17A. This may lead to symptoms such as itching, pain, scaling in psoriasis, swollen and tender joints in psoriatic arthritis, and pain in the spine in axial spondyloarthritis.
COSENTYX contains the active substance secukinumab. This ingredient is a fully-human monoclonal antibody and belongs to a group of medicines called interleukin (IL) inhibitors. Monoclonal antibodies are proteins that recognise and bind specifically to certain proteins in the body.
COSENTYX works by neutralising the activity of the IL-17A protein, which is present at increased levels in conditions such as psoriasis, psoriatic arthritis, and axial spondyloarthritis. It will reduce the inflammation and other symptoms of the condition.
If you have any questions about how COSENTYX works or why this medicine has been prescribed for you, ask your doctor, pharmacist or healthcare provider.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
COSENTYX is not recommended for children and adolescents (under 18 years of age) because it has not been studied in this age group.
COSENTYX may be used by people aged 65 years and over.
COSENTYX is available only with a doctor's prescription and is not addictive.
Before you have COSENTYX
When you must not take it
Do not take COSENTYX if you have had a severe allergic reaction to secukinumab or any of the other ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take COSENTYX if you have an active infection which your doctor thinks is important.
Do not take COSENTYX if there are visible signs of deterioration.
If you think you may be allergic, ask your doctor for advice before using COSENTYX.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to have it
If any of these apply to you, tell your doctor or pharmacist before using COSENTYX:
- you currently have an infection or if you have long-term or repeated infections
- you have tuberculosis
- you have inflammatory bowel disease (Crohn's disease or ulcerative colitis)
- you had a recent vaccination or if you will receive a live vaccine during treatment with COSENTYX
Tell your doctor if you are pregnant, think that you may be pregnant or are planning to have a baby. COSENTYX is not recommended during pregnancy unless the benefits clearly outweigh the potential risks.
Tell your doctor if you are breast-feeding or plan to breast-feed.
If you have not told your doctor about any of the above, tell them before you start taking COSENTYX.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Tell your doctor or pharmacist if you:
- are taking a blood thinning medicine called warfarin.
- are taking, have recently taken or might take any other medicines.
- have recently had or are going to have a vaccination. You should not receive certain types of vaccines (live vaccines) while using COSENTYX.
Such medicines may be affected by COSENTYX or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How COSENTYX is given
Follow all directions given to you by your doctor, nurse or pharmacist carefully. They may differ from the information contained in this leaflet.
Always use COSENTYX as your doctor has told you. You should check with your doctor, nurse or pharmacist if you are not sure.
COSENTYX is intended for subcutaneous use. This means that it is injected into the fatty tissue just under the skin.
The injection may be given by your doctor or nurse or you may be taught how to inject yourself with the medicine.
You and your doctor should decide if you should inject COSENTYX yourself. It is important not to try to inject yourself until you have been trained by your doctor, nurse or pharmacist. A caregiver may also give you your COSENTYX injection after proper training.
If you do not understand the instructions on the label ask your doctor or pharmacist for help.
How much is given
Your doctor will decide how much COSENTYX you need.
Plaque psoriasis
The recommended dose is 300 mg by subcutaneous injection with initial loading dose at Weeks 0, 1, 2, 3, and 4 followed by the same dose every month. Each 300 mg dose is given as two sub-cutaneous injections of 150 mg.
Psoriatic arthritis
The recommended dose is 150 mg by subcutaneous injection with initial dosing at Weeks 0, 1, 2, 3, and 4 followed by the same dose every month. Based on your response, your doctor may increase the dose to 300 mg.
For patients who did not respond well to medicines called tumour necrosis factor (TNF) blockers or patient who also have moderate to severe plaque psoriasis, the recommended dose is 300 mg by subcutaneous injection with initial dosing at Weeks 0, 1, 2, 3, and 4 followed by the same dose every month. Each 300 mg dose is given as two subcutaneous injections of 150 mg.
COSENTYX may be administered with or without methotrexate.
Axial Spondyloarthritis
Ankylosing spondylitis
The recommended dose is 150 mg by subcutaneous injection with initial dosing at Weeks 0, 1, 2, 3, and 4 followed by the same dose every month. Based on your response, your doctor may increase the dose to 300 mg. Each 300 mg dose is given as two subcutaneous injections of 150 mg.
Non-radiographic axial spondyloarthritis
With a loading dose: The recommended dose is 150 mg by subcutaneous injection with initial dosing at Weeks 0, 1, 2, 3, and 4 followed by the same dose every month.
Without a loading dose: The recommended dose is 150 mg by subcutaneous injection every month.
Do not exceed the recommended dose.
Choosing the injection sites
The injection sites are where the skin will be pierced to administer the subcutaneous injection.

The recommended site is the front of your thighs. You may also use the lower abdomen, but not the area five centimetres around the navel (belly button). If a caregiver is giving you the injection, the outer upper arms may also be used (Shown in grey in the diagram for illustrative purposes).

Choose a different site each time you give yourself an injection.
Do not inject into areas where the skin is tender, bruised, red, scaly or hard.
Avoid areas with scars or stretch marks
Injecting COSENTYX yourself
Discuss with your doctor whether or not you will inject COSENTYX yourself.
Do not try to inject yourself if you have not been properly trained or if you are not sure how to do it.
After proper training in injection technique, your doctor will tell you that you may inject it yourself.

Read the following instructions on how to use the prefilled pen all the way through before removing COSENTYX from the refrigerator.
These instructions are to help you to inject correctly.
How to use the prefilled pens
Read ALL the way through these instructions before injecting. These instructions will help you to inject correctly using the pen.
For a more comfortable injection, take the carton containing two pens out of the refrigerator 15 to 30 minutes before injecting.
This will enable them to reach room temperature. COSENTYX should be administered within one hour after removal from 2°C to 8°C storage.
During the injection you will hear 2 loud clicks. The first click indicates that the injection has started. Several seconds later a second click will indicate that the injection is almost finished.
You must keep holding the pen firmly against your skin until you see a green indicator fill the window and stop moving.
Before your injection, gather together the necessary items not included in the COSENTYX pack:
- alcohol swabs
- clean, dry cotton swab or gauze
- sharps disposal container

- When you are ready to use the pens, wash your hands thoroughly with soap and water.
- Inspect the pen.
The liquid should be clear. Its colour may vary from colourless to slightly yellow. You may see a small air bubble, which is normal.
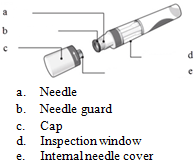
Do NOT use the pen if the liquid contains easily visible particles, is cloudy or is distinctly brown.
- Find a clean, comfortable area.
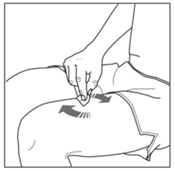
Using a circular motion, clean the injection site with the alcohol swab.
Leave it to dry before injecting. Do not touch the cleaned area again before injecting.
- Only remove the cap when you are ready to use the pen.
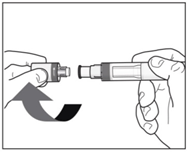
Twist off the cap in the direction of the arrows. Once removed, throw away the cap.
Use the pen within 5 minutes of removing the cap. Do not try to re-attach the cap.
- Hold the pen at 90 degrees to the cleaned injection site.
Ensure the inspection window on the pen is visible before injecting.
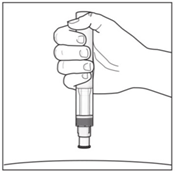
- Press the pen firmly against the skin to start the injection.
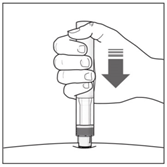
The first click indicates the injection has started.
Keep holding the pen firmly against your skin.
The green indicator shows the progress of the injection.
- Listen for the second click.
This indicates the injection is almost complete.
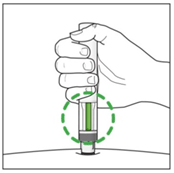
After the injection, check the green indicator fills the window and has stopped moving. The pen can now be removed.
- Check the green indicator fills the window.
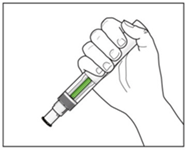
This means the medicine has been delivered.
Contact your doctor if the green indicator is not visible.
There may be a small amount of blood at the injection site.
You can press a cotton ball or gauze over the injection site and hold it for 10 seconds.
Do not rub the injection site.
You may cover the injection site with a small adhesive bandage, if needed.
- Dispose of the used pen in a sharps disposal container (i.e. a puncture-resistant closable container, or similar). Never try to reuse the spent pen.

- If you have been prescribed a 300 mg dose and therefore a second injection is required, choose a different site and repeat steps 2 to 9 with the second pen.
How long to take COSENTYX
This is a long-term treatment. Keep taking this medicine for as long as your doctor tells you.
Your doctor will regularly monitor your condition to check that the treatment is having the desired effect.
If you use more COSENTYX than you should (overdose)
If you or anyone else accidentally received more COSENTYX than you should or sooner than according to the doctor's prescription, immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital.
Do this even if there are no obvious signs of discomfort or poisoning.
If you forget to have it
If you have forgotten to inject a dose of COSENTYX, inject the next dose as soon as you remember. Then talk to your doctor to discuss when you should inject the next dose.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to have your medicine, ask your pharmacist for some hints.
While you are taking COSENTYX
Things you must do
Discontinue treatment and tell your doctor or pharmacist immediately if you get any of these symptoms during treatment with COSENTYX.
Signs or symptoms of a potentially serious infection. These may include:
- fever, flu-like symptoms, night sweats
- feeling tired or short of breath; cough which will not go away
- warm, red and painful skin, or a painful skin rash with blisters
- burning when passing urine
Signs or symptoms of an allergic reaction. These may include:
- difficulty breathing or swallowing
- low blood pressure, which can cause dizziness or light-headedness
- swelling of the face, lips, mouth or throat
- severe itching of the skin, with a red rash or raised bumps
Keep all of your doctor's appointments so that your progress can be checked. Your doctor will do tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Your doctor will decide if and when you may restart the treatment.
If you need to be vaccinated, tell your doctor you are taking COSENTYX before you have the vaccination. Live vaccines will not be suitable for you.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking COSENTYX.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
Things you must not do
Never leave the prefilled pen lying around where others might tamper with it.
Do not open the sealed box until you are ready to use COSENTYX.
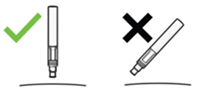
Do not use this medicine if the liquid contains easily visible particles, is cloudy or is distinctly brown.
Do not use the prefilled pen if either the seal on the outer box or the seal of the blister are broken. It may not be safe for you to use.
Do not shake the prefilled pen.
Do not use the pen if it has been dropped with the cap removed.
Do not take it to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Things to be careful of
Dispose of the used COSENTYX prefilled pens immediately after use in a sharps container. The COSENTYX prefilled pen should never be re-used.
If you stop using COSENTYX
Do not stop taking your medicine or change dosage without checking with your doctor first.
Side effects
As with all medicines, patients treated with COSENTYX may experience side effects, although not everybody gets them.
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking COSENTYX.
All medicines can have side effects. Sometimes they are serious, most of the time they are not.
You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Some side effects are very common (These side effects may affect more than 1 in every 10 people)
- Upper respiratory tract infections with symptoms such as sore throat and stuffy nose (nasopharyngitis, rhinitis)
Some side effects are common (These side effects may affect up to 1 in every 10 people)
- Cold sores (oral herpes)
- Diarrhoea
- Runny nose (rhinorrhoea)
- Hives
- Headache
- Nausea
- Itchy rash (urticaria)
Some side effects are uncommon (These side effects may affect up to 1 in every 100 people)
- Oral thrush (oral candidiasis)
- Signs of low levels of white blood cells such as fever, sore throat or mouth ulcers due to infections (neutropenia)
- Athlete's foot (tinea pedis)
- Infection of the external ear (otitis externa)
- Discharge from the eye with itching, redness and swelling (conjunctivitis)
- Painful period
- Some side effects are rare (may affect in to 1 in 1,000 people)
- Severe allergic reaction with shock (anaphylactic shock)
The frequency of some side effects cannot be estimated from available data
- Fungal infections of the skin and mucous membranes (thrush)
New cases of inflammatory bowel disease or "flare-ups" can happen with COSENTYX, and can sometimes be serious. If you have inflammatory bowel disease (ulcerative colitis or Crohn's disease), tell your doctor if you have worsening symptoms during treatment with COSENTYX or develop new symptoms of stomach pain or diarrhoea.
COSENTYX may affect the results of some blood tests, including:
- slight increase in blood cholesterol and blood fat (triglycerides)
- elevated liver enzymes
In clinical trials, major adverse cardiovascular events were rarely observed with COSENTYX.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed here or not yet known may happen in some people.
After taking COSENTYX
Storage
If you have to store COSENTYX:
- Keep it in a refrigerator at 2°C to 8°C out of the sight and reach of children
- Do not freeze it
- Store it in the carton in order to protect it from light.
- If necessary, COSENTYX can be left out of the refrigerator for a single period of up to 4 days at room temperature, not above 30°C. Do not put back in the refrigerator after it has reached room temperature. Discard any unused product.
Do not store COSENTYX or any other medicine in the bathroom, near a sink, or leave it in the car or on a window sill.
A locked section of the refrigerator, at least one-and-a-half metres above the ground, is a good place to store COSENTYX.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
COSENTYX solution for injection is a clear liquid. Its colour may vary from colourless to slightly yellow.
COSENTYX is available in packs containing 1 or 2 single-use pre-filled pens.
Ingredients
Each COSENTYX pre-filled pen contains 150 mg secukinumab as the active substance.
The other ingredients are:
- trehalose dihydrate
- histidine
- histidine hydrochloride monohydrate
- methionine
- polysorbate 80
- water for injection.
This medicine does not contain sucrose, lactose, gluten, tartrazine or any other azo dyes.
Sponsor
COSENTYX is supplied in Australia by:
Novartis Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
Macquarie Park NSW 2113
Telephone 1 800 671 203
® = Registered Trademark
This leaflet was prepared in
September 2020.
Australian Registration Numbers:
Pre-filled pen AUST R 218800
(cos100920c based on PI cos100920i)
Published by MIMS November 2020


 Adverse reactions that occurred at less than 1% frequency in the placebo controlled period of the ERASURE, FIXTURE, FEATURE and JUNCTURE studies through week 12 are given in Table 4.
Adverse reactions that occurred at less than 1% frequency in the placebo controlled period of the ERASURE, FIXTURE, FEATURE and JUNCTURE studies through week 12 are given in Table 4.





 An additional psoriasis study (CLEAR) evaluated 676 patients. Secukinumab 300 mg met the primary and secondary endpoints by showing superiority to ustekinumab based on PASI 90 response at Week 16 and speed of onset of PASI 75 response at Week 4, and long term PASI 90 response at Week 52. Greater efficacy of secukinumab compared to ustekinumab for the endpoints PASI 75/90/100 and IGA mod 2011 0 or 1 response ("clear" or "almost clear") was observed early and continued through to Week 52. In this study, each 300 mg dose was administered as two injections of 150 mg. See Table 10.
An additional psoriasis study (CLEAR) evaluated 676 patients. Secukinumab 300 mg met the primary and secondary endpoints by showing superiority to ustekinumab based on PASI 90 response at Week 16 and speed of onset of PASI 75 response at Week 4, and long term PASI 90 response at Week 52. Greater efficacy of secukinumab compared to ustekinumab for the endpoints PASI 75/90/100 and IGA mod 2011 0 or 1 response ("clear" or "almost clear") was observed early and continued through to Week 52. In this study, each 300 mg dose was administered as two injections of 150 mg. See Table 10. All plaque psoriasis phase III studies included approximately 15 to 25% of patients with concurrent psoriatic arthritis at baseline. Improvements in PASI 75 in this patient population were similar to those in the overall plaque psoriasis population.
All plaque psoriasis phase III studies included approximately 15 to 25% of patients with concurrent psoriatic arthritis at baseline. Improvements in PASI 75 in this patient population were similar to those in the overall plaque psoriasis population. The safety profiles of the two dosing regimens, Cosentyx 300 mg administered every 4 weeks and Cosentyx 300 mg administered every 2 weeks, in patients weighing ≥ 90 kg were comparable and consistent with the safety profile reported in psoriasis patients.
The safety profiles of the two dosing regimens, Cosentyx 300 mg administered every 4 weeks and Cosentyx 300 mg administered every 2 weeks, in patients weighing ≥ 90 kg were comparable and consistent with the safety profile reported in psoriasis patients.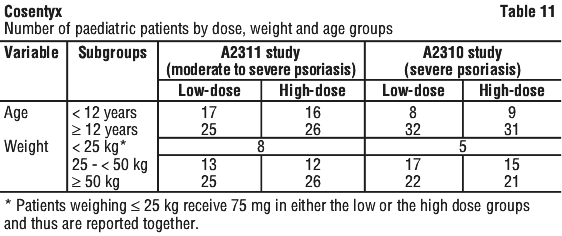
 A higher proportion of paediatric patients treated with secukinumab reported improvement in health-related quality of life as measured by a CDLQI score of 0 or 1 compared to placebo at Week 12 (low dose 44.7%, high dose 50%, placebo 15%). This improvement was further maintained through Week 52.
A higher proportion of paediatric patients treated with secukinumab reported improvement in health-related quality of life as measured by a CDLQI score of 0 or 1 compared to placebo at Week 12 (low dose 44.7%, high dose 50%, placebo 15%). This improvement was further maintained through Week 52.
 The onset of action of Cosentyx occurred as early as Week 2. Statistically significant difference in ACR 20 vs placebo was reached at Week 3. In PsA2 efficacy responses were maintained up to Week 104. At Week 16, Cosentyx treated patients demonstrated significant improvements in signs and symptoms among which significantly higher responses in ACR 20 (60.0% and 57.0% for 150 mg and 300 mg, respectively) compared to placebo (18.4%).
The onset of action of Cosentyx occurred as early as Week 2. Statistically significant difference in ACR 20 vs placebo was reached at Week 3. In PsA2 efficacy responses were maintained up to Week 104. At Week 16, Cosentyx treated patients demonstrated significant improvements in signs and symptoms among which significantly higher responses in ACR 20 (60.0% and 57.0% for 150 mg and 300 mg, respectively) compared to placebo (18.4%). Similar responses for primary and key secondary endpoints were seen in PsA patients regardless of whether they were on concomitant MTX treatment or not.
Similar responses for primary and key secondary endpoints were seen in PsA patients regardless of whether they were on concomitant MTX treatment or not. Inhibition of structural damage was maintained with Cosentyx 150 mg treatment up to Week 104 in PsA2 study and with Cosentyx 300 mg treatment up to week 52 in PsA3 study.
Inhibition of structural damage was maintained with Cosentyx 150 mg treatment up to Week 104 in PsA2 study and with Cosentyx 300 mg treatment up to week 52 in PsA3 study. Improvement in ASAS 20 for both secukinumab doses were observed by Week 4 and were maintained up to 52 weeks.
Improvement in ASAS 20 for both secukinumab doses were observed by Week 4 and were maintained up to 52 weeks.
 The onset of action of Cosentyx 150 mg occurred as early as Week 1 for ASAS 20 and Week 2 for ASAS 40 (superior to placebo) in AS2 study. (See Figures 4 and 5).
The onset of action of Cosentyx 150 mg occurred as early as Week 1 for ASAS 20 and Week 2 for ASAS 40 (superior to placebo) in AS2 study. (See Figures 4 and 5).
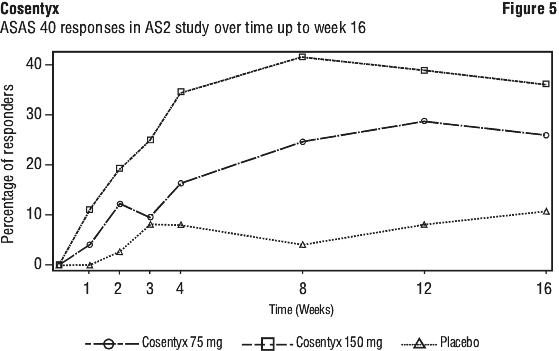 ASAS 20 responses were improved at Week 16 in both anti-TNF-α naïve patients (68.2% vs. 31.1%; p < 0.05) and anti-TNF-α-IR patients (50.0% vs. 24.1%; p < 0.05) for Cosentyx 150 mg compared with placebo, respectively.
ASAS 20 responses were improved at Week 16 in both anti-TNF-α naïve patients (68.2% vs. 31.1%; p < 0.05) and anti-TNF-α-IR patients (50.0% vs. 24.1%; p < 0.05) for Cosentyx 150 mg compared with placebo, respectively. The onset of action of Cosentyx 150 mg occurred as early as Week 3 for ASAS 40 in anti-TNF-alpha naive patients (superior to placebo) in nr-axSpA1 study. The percentage of patients achieving an ASAS 40 response in anti-TNF-alpha naive patients by visit is shown in Figure 6. Patients treated with Cosentyx maintained their response compared to placebo up to Week 52.
The onset of action of Cosentyx 150 mg occurred as early as Week 3 for ASAS 40 in anti-TNF-alpha naive patients (superior to placebo) in nr-axSpA1 study. The percentage of patients achieving an ASAS 40 response in anti-TNF-alpha naive patients by visit is shown in Figure 6. Patients treated with Cosentyx maintained their response compared to placebo up to Week 52. ASAS 40 responses were also improved at Week 16 in anti-TNF-alpha-IR patients (28.6% vs. 13.3%) for Cosentyx 150 mg compared with placebo. The magnitude of response (treatment difference versus placebo) with respect to signs and symptoms at Week 16 was similar in anti-TNF-alpha-naïve and anti-TNF-alpha-IR patients, with higher absolute response rates in anti-TNF-alpha-naïve patients. Efficacy versus placebo was maintained in anti-TNF-alpha-naïve and anti-TNF-alpha-IR patients up to Week 52.
ASAS 40 responses were also improved at Week 16 in anti-TNF-alpha-IR patients (28.6% vs. 13.3%) for Cosentyx 150 mg compared with placebo. The magnitude of response (treatment difference versus placebo) with respect to signs and symptoms at Week 16 was similar in anti-TNF-alpha-naïve and anti-TNF-alpha-IR patients, with higher absolute response rates in anti-TNF-alpha-naïve patients. Efficacy versus placebo was maintained in anti-TNF-alpha-naïve and anti-TNF-alpha-IR patients up to Week 52.

 In both studies, the onset of action of secukinumab occurred as early as Week 2, the efficacy progressively increased to Week 16 and was maintained up to Week 52.
In both studies, the onset of action of secukinumab occurred as early as Week 2, the efficacy progressively increased to Week 16 and was maintained up to Week 52.
