What is in this leaflet
This leaflet answers some common questions about CYRAMZA. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking CYRAMZA against the benefits this medicine is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may need to read it again.
The information in this leaflet was last updated on the date shown on the final page. More recent information on this medicine may be available. Make sure you speak to your pharmacist, nurse or doctor to obtain the most up to date information on this medicine. You can also download the most up to date leaflet from www.lilly.com.au. The updated leaflet may contain important information about CYRAMZA and its use that you should be aware of.
What CYRAMZA is used for
CYRAMZA contains the active ingredient ramucirumab.
CYRAMZA is used to treat advanced gastric cancer (including cancer of the junction between the oesophagus and the stomach).
It belongs to a group of medicines known as antineoplastic (anti-cancer) agents. They may also be called chemotherapy medicines.
It works by cutting off the blood supply that allows cancer cells to grow.
Your doctor may have prescribed it for another reason.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
This medicine is available only with a doctor's prescription.
CYRAMZA may be used in combination with other chemotherapy drugs.
Before you are given CYRAMZA
When you must not be given it
Do not take CYRAMZA if you have an allergy to:
- any medicine containing ramucirumab
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
If you are not sure whether you should be given this medicine, talk to your doctor.
Before you are given it
Tell your doctor if you have or have had any of the following medical conditions:
- any condition which increases the risk of bleeding, or you are taking any medicines which may increase the risk of bleeding or which affect blood clotting ability.
- high blood pressure. Your doctor will make sure that your blood pressure is brought under control before starting CYRAMZA.
- severe liver disease ('cirrhosis') and associated conditions, such as excessive accumulation of fluid in your abdomen ('ascites').
- are going to have planned surgery, if you have had recent surgery or if you have a poorly healing wound after surgery. CYRAMZA may increase the risk of problems with wound healing. You should not receive CYRAMZA for at least 4 weeks before you undergo planned surgery and your doctor will decide when to start treatment.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Pregnancy and breast-feeding should be avoided during CYRAMZA treatment and for at least 3 months after the last dose of CYRAMZA. Your doctor can discuss with you the risks and benefits involved.
This medicine is not recommended for use in children under the age of 18 years. Safety and effectiveness in children younger than 18 years have not been established.
If you have not told your doctor about any of the above, tell him/her before you take CYRAMZA.
Taking Premedication
Your doctor may advise you to take certain medicines to reduce the risk of an infusion-related reaction before you receive CYRAMZA.
If you experience an infusion-related reaction during CYRAMZA therapy, you will be given premedication for all future infusion.
Ask your doctor if you have any questions about why these other medicines have been prescribed for you.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines maybe affected by CYRAMZA.
You may need different amounts of your medicines or to stop taking them for a few days. Or you may need to take different medicines.
How CYRAMZA will be given
Follow all directions given to you by your doctor or pharmacist carefully.
How much is given
Your doctor will decide the dosage of CYRAMZA you should take. This will depend on your body weight.
How it is given
CYRAMZA is given as an infusion (drip) into your veins over approximately 60 minutes.
You may also be given other chemotherapy medicines.
If you experience an infusion-related reaction during treatment, the time taken to give your infusion will be increased for the duration of that infusion and all future infusions.
Your doctor or nurse will inject CYRAMZA for you.
Never inject CYRAMZA yourself. Always let your doctor or nurse do this.
How often it is given
CYRAMZA is given once every two weeks as part of the treatment cycle. Your doctor will advise how many treatment cycles you need.
CYRAMZA treatment will be temporarily stopped if you:
- develop severe high blood pressure, until it is controlled with anti-hypertensive medication
- develop wound healing problems, until the wound is healed or prior to planned surgery.
The amount of protein in your urine will be checked regularly during treatment. Your doctor may decide to change your dose or delay treating you depending on your general condition and if your urine protein levels are too high. Once your urine protein level has decreased to a certain level, treatment may be restarted with a lower dose.
CYRAMZA treatment will be permanently stopped if you:
- develop a blood clot in your arteries
- develop a hole in your gut wall
- experience severe bleeding
- experience a severe infusion-related reaction
- develop high blood pressure that cannot be controlled with medication
- are passing more than a certain amount of protein with your urine or if you develop a severe kidney disease (nephrotic syndrome)
- notice any changes in your movement or behaviour
- develop abnormal tube-like connections or passageways inside the body between internal organs and skin or other tissues
Overdose
As CYRAMZA therapy is given to you under the supervision of your doctor or nurse, it is unlikely that you will have too much.
However, if you experience any unexpected or worrying side effects after being given CYRAMZA, immediately tell your doctor or nurse or go to the Emergency Department at your nearest hospital. You may need urgent medical attention.
While you are receiving CYRAMZA
Things you must do
Talk to your doctor or nurse immediately if any of the following applies to you (or you are not sure) during treatment with CYRAMZA or thereafter:
- blocking of the arteries by a blood clot. CYRAMZA can cause blood clots in your arteries. Arterial blood clots can lead to serious conditions, including heart attack or stroke. Symptoms of a heart attack may include chest pain or heaviness in the chest. Symptoms of a stroke may include sudden numbness or weakness of the arm, leg and face, feeling confused, difficulty speaking or understanding others, sudden difficulty in walking or loss of balance or coordination, or sudden dizziness. CYRAMZA will be permanently stopped if you develop a blood clot in your arteries.
- holes in your gut wall. CYRAMZA has the potential to increase the risk of holes in your gut wall. Symptoms may include severe abdominal pain, being sick (vomiting), fever or chills. CYRAMZA will be permanently stopped if you develop a hole in your gut wall.
- severe bleeding. CYRAMZA has the potential to cause severe bleeding. Symptoms may include extreme tiredness, weakness, dizziness or changes in the colour of your stools. CYRAMZA will be permanently stopped if you experience severe bleeding.
- infusion-related reactions. Infusion-related reactions may happen with treatment with CYRAMZA. Your doctor or nurse will check for side effects during your infusion. Symptoms may include increased muscle tension and/or tremors, back pain and/or spasms, chest pain and/or tightness, chills, flushing, difficulty breathing, wheezing, and feeling of tingling or numbness in the hands or feet. In severe cases, symptoms may include breathing distress caused by narrowing of the airways, faster heartbeat, and feeling faint. CYRAMZA will be permanently stopped if you experience a severe infusion-related reaction.
- abnormal tube-like connections or passageways inside the body. CYRAMZA may increase the risk of abnormal tube-like connections or passageways inside the body between internal organs and skin or other tissues. Cyramza will be permanently stopped if you develop fistulae.
Tell your partner or caregiver you are receiving CYRAMZA and ask them to tell you if they notice any changes in your movement or behaviour. If they notice any changes you should tell your doctor about them immediately.
Talk to your doctor about regular blood pressure monitoring. CYRAMZA may cause high blood pressure. Most people have no symptoms from this. Talk to your doctor about measuring your blood pressure. CYRAMZA will be permanently stopped if you develop high blood pressure that cannot be managed with medication.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are receiving CYRAMZA.
Tell any other doctors, dentists and pharmacists who treat you that you are receiving this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are receiving this medicine. It may increase the risk of problems with wound healing. You should not receive CYRAMZA before you undergo planned surgery and your doctor will decide when to re-start treatment.
If you become pregnant while receiving this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are receiving this medicine.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things to be careful of
Be careful driving or operating machinery until you know how CYRAMZA affects you. It is not known whether CYRAMZA will affect your ability to drive or to use machines. If you experience any symptoms affecting your ability to concentrate and react, do not drive, operate or do anything else that could be dangerous until the effect goes away.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking CYRAMZA.
This medicine is to help people with gastric cancer, including cancers of the junction between the oesophagus and the stomach, but it may have unwanted side effects in some people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Because CYRAMZA may be used with other medicines that treat cancer, it may be difficult for your doctor to tell whether the side effects are due to CYRAMZA or due to other medicines.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- feeling tired or weak
- diarrhoea
- nose bleed
- abdominal pain
- intestinal blockage; symptoms may include constipation and abdominal pain
- swelling of the hands, feet and legs due to fluid retention
- high blood pressure
- inflammation of the mouth
- headache
- muscle weakness or twitching
- abnormal heart rhythm
- confusion
- bleeding or bruising more easily than normal
- low blood levels of potassium which can cause muscle weakness, twitching or abnormal heart rhythm
- low blood levels of sodium which can cause tiredness and confusion or muscle twitching.
- low blood levels of thyroid hormone which can cause tiredness, weight gain, and feeling cold.
- Changes in speech
The above lists include the more common side effects of your medicine. When used in combination with other chemotherapy medicine, also refer to the other product's consumer medicine information leaflet for a list of other possible side effects.
Tell your doctor as soon as possible if you notice any of the following:
- fever or infection with a temperature, sweating or other signs of infection
- severe abdominal pain
- being sick (vomiting)
- extreme tiredness, weakness or dizziness
- changes in the colour of your stools.
The above list includes serious side effects which may require medical attention.
If any of the following happen, tell your doctor immediately or go to the Emergency Department at your nearest hospital:
- signs of a heart attack such as chest pain or heaviness in the chest
- signs of a stroke such as sudden numbness or weakness of the arm, leg and face, feeling confused, difficulty speaking or understanding others, sudden difficulty in walking or loss of balance or coordination, or sudden dizziness
- sudden signs of allergy such as rash, itching or hives on the skin, swelling of the face, lips or tongue or other parts of the body, shortness of breath, wheezing or trouble breathing.
- vision loss associated with headaches, confusion and seizures
- one or a combination of the following: confusion, disorientation or memory loss, changes in the way you move, walk or talk, decreased strength or progressive weakness in your body, blurred or loss of vision.
The above list includes very serious side effects and can be life-threatening. You may need urgent medical attention or hospitalisation. Serious side effects are rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people. Some of these side effects (for example, abnormal blood tests showing low cell counts, low blood levels of albumin, potassium or sodium, or urine tests showing high protein levels) can only be found when your doctor does tests to check your progress.
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
After receiving CYRAMZA
Storage
This medicine will be stored in the hospital pharmacy or on the ward.
It will be kept in a refrigerator at a temperature between 2°C and 8°C in the outer carton to protect from light.
Disposal
CYRAMZA is for single use only.
The vials should be used once only and any remaining contents should be discarded.
Product description
What it looks like
CYRAMZA is a clear to slightly opalescent and colourless to slightly yellow solution and is available in a glass vial container with a rubber stopper.
CYRAMZA is available in packs of:
- 1 vial of 10 mL
- 1 vial of 50 mL.
Not all pack sizes may be marketed.
Ingredients
CYRAMZA is supplied in 10 mL and 50 mL vials.
The 10 mL vial of CYRAMZA contains 100 mg of ramucirumab, 6.5 mg of histidine, 12.2 mg of histidine hydrochloride monohydrate, 99.8 mg of glycine, 43.8 mg of sodium chloride, 1 mg of polysorbate 80 and water for injections to 10mL.
The 50 mL vial of CYRAMZA contains 500 mg of ramucirumab, 32.5 mg of histidine, 61 mg of histidine hydrochloride monohydrate, 499 mg of glycine, 219 mg of sodium chloride, 5 mg of polysorbate 80 and water for injections to 50mL.
Supplier
CYRAMZA is supplied in Australia by:
Eli Lilly Australia Pty Limited
112 Wharf Road
WEST RYDE NSW 2114
Australian Registration Numbers:
CYRAMZA 100 mg, AUST R 227351
CYRAMZA 500 mg, AUST R 227352
This leaflet was prepared in December 2020.
®= Registered Trademark
Published by MIMS January 2021

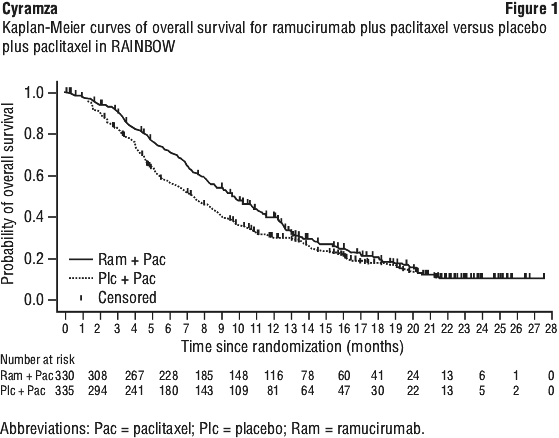
 Clinically relevant ADRs reported in ≥ 1% and < 5% of the ramucirumab plus paclitaxel treated patients in RAINBOW were gastrointestinal perforation (1.2% ramucirumab plus paclitaxel versus 0.3% for placebo plus paclitaxel) and sepsis (3.1% ramucirumab plus paclitaxel versus 1.8% placebo plus paclitaxel).
Clinically relevant ADRs reported in ≥ 1% and < 5% of the ramucirumab plus paclitaxel treated patients in RAINBOW were gastrointestinal perforation (1.2% ramucirumab plus paclitaxel versus 0.3% for placebo plus paclitaxel) and sepsis (3.1% ramucirumab plus paclitaxel versus 1.8% placebo plus paclitaxel). Clinically relevant ADRs reported in ≥ 1% and < 5% of the ramucirumab treated patients in REGARD were: neutropenia, arterial thromboembolic events, intestinal obstruction, epistaxis, and rash.
Clinically relevant ADRs reported in ≥ 1% and < 5% of the ramucirumab treated patients in REGARD were: neutropenia, arterial thromboembolic events, intestinal obstruction, epistaxis, and rash.




 The average molecular mass of ramucirumab with the predominant form of N-linked glycosylation is 146,756 Da.
The average molecular mass of ramucirumab with the predominant form of N-linked glycosylation is 146,756 Da.