What is in this leaflet
This leaflet answers some common questions about DBL Docetaxel, Concentrated Injection.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking DBL Docetaxel, Concentrated Injection against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What DBL Docetaxel, Concentrated Injection is used for
Docetaxel is used to treat:
- breast cancer
- non small cell lung cancer
- ovarian cancer
- prostate cancer
- head and neck cancer
This medicine belongs to a group of medicines called antineoplastic or cytotoxic medicines. You may also hear of these being called chemotherapy medicines.
It works by stopping cells from growing and multiplying.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
This medicine is available only with a doctor’s prescription.
You may have taken another medicine to treat your breast, non small cell lung cancer, ovarian, prostate or head and neck cancer. However, your doctor has now decided to treat you with docetaxel.
There is not enough information to recommend the use of this medicine for children.
Before you are given DBL Docetaxel, Concentrated Injection
When you must not be given it
Do not use DBL Docetaxel, Concentrated Injection if you have an allergy to:
- any medicine containing docetaxel or polysorbate 80
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing or a tight feeling in your chest
- swelling of the face, lips, tongue or other parts of the body
- rash, itching, hives or flushed, red skin
- dizziness or light-headedness
- back pain
Do not use DBL Docetaxel, Concentrated Injection if you have, or have had, any of the following medical conditions:
- severe liver problems
- blood disorder with a reduced number of white blood cells
Females: Do not use this medicine if you are pregnant or intend to become pregnant.
If you become pregnant during treatment or think you might be pregnant, speak to your doctor as soon as possible.
You should use a reliable method of contraception during treatment with DBL Docetaxel Injection and for at least 2 months after your last dose.
Like most medicines used to treat cancer, docetaxel is not recommended for use during pregnancy, unless you and your doctor have discussed the risks and benefits involved.
Do not breast-feed during treatment with this medicine and for 1 week after your last dose. It is not known if docetaxel passes into breast milk and there is a possibility that your baby may be affected.
Males: You should use a reliable method of contraception during treatment with DBL Docetaxel Injection and for at least 4 months after your last dose, if you are with a female partner who is able to become pregnant.
You should seek advice from your doctor on preserving sperm before starting treatment.
Both males and females should discuss the need for individual genetic counselling with their doctor if you decide to have a child after treatment.
Docetaxel may alter fertility in males and females.
If you are not sure whether you should start using this medicine, talk to your doctor.
Before you are given it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Docetaxel is not recommended for use in children. Safety and effectiveness in children have not been established.
Tell your doctor if you have or have had any of the following medical conditions:
- blood disorder with a reduced number of white blood cells
- hearing problems
- heart problems
- high uric acid levels
- kidney problems
- liver problems
Tell your doctor if you have an infection or high temperature. Your doctor may decide to delay your treatment until the infection has gone. A mild illness, such as a cold, is not usually a reason to delay treatment.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell him/her before you start using DBL Docetaxel, Concentrated Injection.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and DBL Docetaxel, Concentrated Injection may interfere with each other. These include:
- other medicines used to treat cancer, radiation therapy or any other treatment which lowers your immune system, including cyclosporin
- some medicines used to treat bacterial infections, including erythromycin, clarithromycin and telithromycin.
- Ketoconazole, itraconazole and voriconazole, medicines used to treat fungal infections
- nifedipine, a medicine used to treat high blood pressure and angina
- some medicines called corticosteroids
- medicines used to treat people with epilepsy, such as phenobarbitone
- medicines used to treat or prevent HIV or Hepatitis C viral infections, such as ritonavir, indinavir, nelfinavir and saquinavir.
- medicines used to treat people with depression such as nefazodone
These medicines may be affected by DBL Docetaxel, Concentrated Injection or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How DBL Docetaxel, Concentrated Injection is given
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
Docetaxel should only be administered by trained professionals, with appropriate handling, in a hospital or clinic environment.
Before you are given your docetaxel infusion your doctor should:
- prescribe you an oral corticosteroid (eg dexamethasone) to help stop or reduce the severity of certain side effects. For breast, lung, ovarian and head and neck cancer, this medicine is usually taken for three days (one day before, the day of and the day after your infusion). These medicines are very important. For prostate cancer, this is usually taken on the day of the infusion (12 hours, 3 hours and 1 hour before your infusion).
- test your blood to see how many white blood cells you have. If they are too low, your infusion may be delayed.
- test your blood for levels of liver enzymes. If these levels are high your doctor may reduce your dose or decide you should not have a docetaxel infusion at that time.
Ask your doctor or pharmacist if you have any questions on these medicines or tests.
How much is given
Your doctor will decide what dose you will receive. This depends on your condition and other factors, such as your weight, kidney function and other chemotherapy medicines you are being given.
The usual dose of docetaxel is 75 to 100mg/m2 which is based on your body size (m2).
When docetaxel is given in combination with capecitabine (another medicine used for the treatment of breast cancer) the usual dose of docetaxel is 75 mg/m2.
Docetaxel may be given alone or in combination with other drugs.
Several courses of docetaxel therapy may be needed depending on your response to treatment.
Additional treatment may not be repeated until your blood cell numbers return to acceptable levels and any uncontrolled effects have been controlled.
Ask your doctor if you want to know more about the dose of docetaxel you receive.
How it is given
DBL Docetaxel, Concentrated Injection is given as an infusion (drip) into your veins, over 1 hour.
How long it is given for
Docetaxel is given every 3 weeks. This is called one cycle of chemotherapy. Your doctor will decide how many of these cycles you will need.
If you take too much (overdose)
As DBL Docetaxel, Concentrated Injection is given to you under the supervision of your doctor, it is very unlikely that you will receive too much. However, if you experience any severe side effects after being given DBL Docetaxel, Concentrated Injection, tell your doctor immediately or go to Accident and Emergency at the nearest hospital. You may need urgent medical attention.
In case of overdose, immediately contact the Poisons Information Centre 13 11 26
While you are using DBL Docetaxel, Concentrated Injection
Things you must do
Keep follow up appointments with your doctor. It is important to have your follow-up doses/cycles/infusions of docetaxel at the appropriate times to get the best effects from your treatments.
Your doctor may want to check your blood pressure and do some blood and other tests from time to time to check on your progress and detect any unwanted side effects.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are about to be started on any new medicine, tell your doctor, dentist or pharmacist that you are having DBL Docetaxel, Concentrated Injection.
If you plan to have surgery that needs a general anaesthetic, tell your doctor or dentist that you are having docetaxel.
If you become pregnant while having docetaxel, tell your doctor immediately.
DBL Docetaxel, Concentrated Injection can lower the number of white blood cells and platelets in your blood. This means that you have an increased chance of getting an infection or bleeding.
The following precautions should be taken to reduce your risk of infection or bleeding:
Avoid people who have infections. Check with your doctor immediately if you think you may be getting an infection, or if you get a fever, chills, cough, hoarse throat, lower back or side pain or find it painful or difficult to urinate.
Be careful when using a toothbrush, toothpick or dental floss. Your doctor, dentist, nurse or pharmacist may recommend other ways to clean your teeth and gums. Check with your doctor before having any dental work.
Be careful not to cut yourself when you are using sharp objects such as a razor or nail cutters.
Avoid contact sports or other situations where you may bruise or get injured.
If you notice swelling in the feet and legs or a slight weight gain, inform your doctor or nurse. Docetaxel may cause fluid retention which means the body is holding extra water. If this fluid retention is in the chest or around the heart it can be life threatening. In most cases, fluid retention will go away within weeks or months after your treatments are completed.
Things you must not do
Do not use DBL Docetaxel, Concentrated Injection to treat any other complaints unless your doctor tells you to.
Things to be careful of
Be careful driving or operating machinery until you know how DBL Docetaxel, Concentrated Injection affects you. Docetaxel may cause dizziness, light-headedness, tiredness, drowsiness in some people. Make sure you know how you react to docetaxel before you drive a car, operate machinery, or do anything else that could be dangerous if you are dizzy or light-headed. If this occurs do not drive. If you drink alcohol, dizziness or light-headedness may be worse.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking DBL Docetaxel, Concentrated Injection.
It helps most people with breast, non small cell lung cancer, ovarian, prostate or head and neck cancer, but it may have unwanted side effects in a few people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
If you are over 60 years of age you may have an increased chance of getting side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- irritation, pain, swelling or colouring around the needle during infusion
- high temperature
- stomach pain or discomfort
- feeling sick, upset stomach or vomiting
- mild diarrhoea
- constipation
- inflammation of the food pipe (oesophagus)
- whitening or darkening of the skin or nails
- loosening of the nails
- unusual hair loss or thinning
- joint pain or swelling
- aching muscles, muscle tenderness, swelling or weakness not caused by exercise
- unusual tiredness or weakness
- mild swelling of hands, ankles and feet
- weight gain/loss
- pins and needles or a burning or tingling feeling in hands or feet
- redness or rash around previous radiation site (if you have had radiotherapy)
- back pain
- decreased appetite
- high blood pressure (hypertension)
- low blood pressure (hypotension)
- temporary visual disturbances, or feeling that you are about to faint, which mostly occur when you are being infused with docetaxel.
The above list includes the more common side effects of your medicine.
Tell your doctor as soon as possible if you notice any of the following:
- infections
- frequent infections with fever, severe chills, sore throat or mouth ulcers - especially 5-7 days after receiving a docetaxel infusion
- sore red mouth or vagina or swelling in these areas
- severe diarrhoea
- tiredness, headaches, being short of breath when exercising, dizziness and looking pale
- coughing
- change in the rhythm or rate of your heart beats (palpitations)
- muscle pain or cramps
- flushed, dry skin, irritability and confusion
- passing little or no urine, drowsiness, nausea, vomiting and breathlessness
- yellowing of the skin or eyes, also called jaundice
- flaking of the skin
- red, scaly patches of the skin especially around the cheeks and nose
- raised lumps on the skin which looks like scalding
- hardening of the skin
- excessive watery discharge from the eyes
- trouble with your hearing, or some loss of hearing
- sudden and severe swelling or pain in the joints or rash.
- Blurry or "wavy" vision or disturbances to colour vision.
- Widening and rounding of the tips of the fingers or toes (clubbing).
- Leg pain, tenderness or swelling.
- Skin that is red and warm.
The above list includes serious side effects that may require medical attention.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- sudden signs of allergy such as rash, itching, hives on the skin, swelling of the face, tongue or other parts of the body, shortness of breath, wheezing or trouble breathing
- Redness of the skin studded with pimples containing fluid or pus
- convulsions, fits or seizures
- hallucinations or delirium
- ulcer in the stomach or intestine - vomiting blood or material that looks like coffee grounds, bleeding from the back passage, black sticky bowel motions or bloody diarrhoea
- difficulty in breathing, rapid and/or shallow breathing
- sudden swelling of the leg/arm which may be due to blood clots
- pain, pressure or tightness in the chest, arms, neck, jaw or back that may occur together with nausea, dizziness or cold sweats
- fainting
- fever with diarrhoea and or vomiting, painful cramping.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
These side effects may differ when using docetaxel in combination with another chemotherapy agent.
Please consult your doctor for possible side effects that may be caused by using docetaxel with another chemotherapy agent.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell. Other side effects not listed above may also occur in some people.
The benefits and side effects of docetaxel may take some time to occur. Therefore, even after you have finished your docetaxel treatment you should tell your doctor immediately if you notice any of the side effects listed in this section.
After using DBL Docetaxel, Concentrated Injection
Storage
DBL Docetaxel, Concentrated Injection will be stored in the pharmacy or on the ward. The injection is kept in a cool dry place, protected from light, where the temperature stays below 25°C.
Product description
What it looks like
DBL Docetaxel, Concentrated Injection in single dose vials is a clear, colourless to pale yellow solution, which is diluted prior to intravenous administration.
Ingredients
Active ingredients:
DBL Docetaxel, Concentrated Injection contains 10mg/mL of docetaxel as the active ingredient.
Inactive Ingredients
It also contains:
- ethanol
- citric acid
- Polysorbate 80
- Macrogol 300
DBL Docetaxel, Concentrated Injection does not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
Sponsor
Pfizer Australia Pty Ltd
Sydney NSW
Toll Free Number: 1800 675 229
www.pfizermedicalinformation.com.au
DBL Docetaxel, Concentrated Injection is available in the following strengths:
- 20 mg/2 mL vials
AUST R 163802 - 80 mg/8mL vials
AUST R 163801 - 160 mg/16mL vials
AUST R 163803
™ = Trademark
This leaflet was prepared in September 2024.
Published by MIMS November 2024

 35 toxic deaths (1.7%) were reported in the 2,045 patients with normal baseline liver function tests treated with docetaxel as monotherapy at the initially planned dose of 100 mg/m2. Septic deaths (neutropenic infections, pneumonia or sepsis) accounted for 80% of the toxic deaths. The incidence of toxic deaths was higher (9.8%) in patients with elevated baseline LFTs.
35 toxic deaths (1.7%) were reported in the 2,045 patients with normal baseline liver function tests treated with docetaxel as monotherapy at the initially planned dose of 100 mg/m2. Septic deaths (neutropenic infections, pneumonia or sepsis) accounted for 80% of the toxic deaths. The incidence of toxic deaths was higher (9.8%) in patients with elevated baseline LFTs. Frequent grade 3 and 4 laboratory abnormalities are shown in Table 4.
Frequent grade 3 and 4 laboratory abnormalities are shown in Table 4. Rare or uncommon adverse effects, as described for capecitabine monotherapy, can be expected for combination therapy as well. See capecitabine product information for adverse effects which are at least remotely related to capecitabine occurring in < 5% of patients treated with capecitabine in combination with docetaxel.
Rare or uncommon adverse effects, as described for capecitabine monotherapy, can be expected for combination therapy as well. See capecitabine product information for adverse effects which are at least remotely related to capecitabine occurring in < 5% of patients treated with capecitabine in combination with docetaxel. There was an increased incidence of SAEs (40% vs. 31%) and grade 4 AEs (34% vs. 23%) in the combination arm compared to docetaxel monotherapy.
There was an increased incidence of SAEs (40% vs. 31%) and grade 4 AEs (34% vs. 23%) in the combination arm compared to docetaxel monotherapy. In this study, all patients had a baseline cardiac ejection fraction of greater than 50%. In the docetaxel plus trastuzumab arm, 64% had received a prior anthracycline as adjuvant therapy, compared with 55% in the docetaxel alone arm.
In this study, all patients had a baseline cardiac ejection fraction of greater than 50%. In the docetaxel plus trastuzumab arm, 64% had received a prior anthracycline as adjuvant therapy, compared with 55% in the docetaxel alone arm. Of the 744 patients treated with TAC (docetaxel, doxorubicin and cyclophosphamide), 33.1% experienced severe TEAEs. Dose reductions due to haematological toxicity occurred in 1% of cycles in TAC arm. 6% of patients treated with TAC discontinued treatment due to adverse events, fever in the absence of infection and allergy being the most common reasons for withdrawal. Two patients died within 30 days of their last study treatment; 1 death was considered to be related to study drug.
Of the 744 patients treated with TAC (docetaxel, doxorubicin and cyclophosphamide), 33.1% experienced severe TEAEs. Dose reductions due to haematological toxicity occurred in 1% of cycles in TAC arm. 6% of patients treated with TAC discontinued treatment due to adverse events, fever in the absence of infection and allergy being the most common reasons for withdrawal. Two patients died within 30 days of their last study treatment; 1 death was considered to be related to study drug. The 3-year cumulative incidence of all symptomatic cardiac events was 2.36% and 1.16% in the AC-TH and TCH arms, respectively (versus 0.52% in the AC-T control arm, see Section 5.1 Pharmacodynamic Properties). The 3-year cumulative incidence of CHF events (grade 3 or 4) was 1.9% and 0.4% in the AC-TH and TCH arms, respectively (versus 0.3% in the AC-T control arm).
The 3-year cumulative incidence of all symptomatic cardiac events was 2.36% and 1.16% in the AC-TH and TCH arms, respectively (versus 0.52% in the AC-T control arm, see Section 5.1 Pharmacodynamic Properties). The 3-year cumulative incidence of CHF events (grade 3 or 4) was 1.9% and 0.4% in the AC-TH and TCH arms, respectively (versus 0.3% in the AC-T control arm).


 The most frequent adverse events reported for docetaxel were neutropenia, febrile neutropenia, gastrointestinal disorders, neurological disorders, asthenia and fluid retention. More grade 3/4 events were observed from docetaxel (55.4%) compared to paclitaxel (23.0%). No unexpected toxicities were reported for docetaxel.
The most frequent adverse events reported for docetaxel were neutropenia, febrile neutropenia, gastrointestinal disorders, neurological disorders, asthenia and fluid retention. More grade 3/4 events were observed from docetaxel (55.4%) compared to paclitaxel (23.0%). No unexpected toxicities were reported for docetaxel.

 The beneficial effect of TAC was seen in both hormone receptor positive and negative patients.
The beneficial effect of TAC was seen in both hormone receptor positive and negative patients. There were 29% of patients with high risk node negative disease included in the study. The benefit observed for the overall population was irrespective of the nodal status. See Table 17.
There were 29% of patients with high risk node negative disease included in the study. The benefit observed for the overall population was irrespective of the nodal status. See Table 17.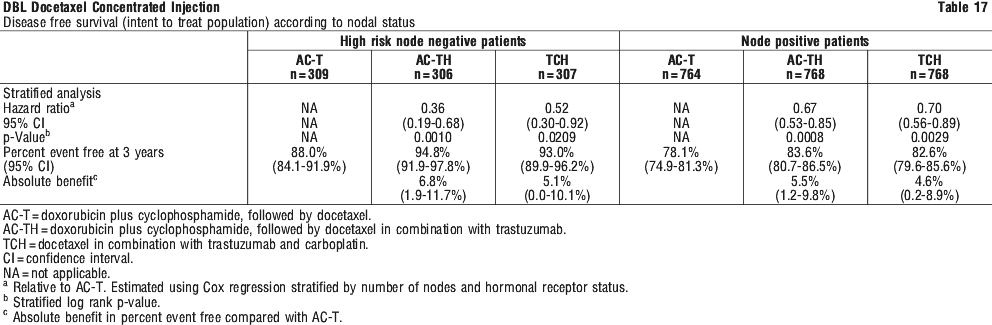



 Chemical name: (2R,3S)-N-carboxy-3-phenylisoserine, N-tert-butyl ester, 13-ester with 5β, 20-epoxy-1,2α,4,7β,10β,13α -hexahydroxytax-11-en-9-one 4- acetate 2-benzoate.
Chemical name: (2R,3S)-N-carboxy-3-phenylisoserine, N-tert-butyl ester, 13-ester with 5β, 20-epoxy-1,2α,4,7β,10β,13α -hexahydroxytax-11-en-9-one 4- acetate 2-benzoate.