1 Name of Medicine
Dexrazoxane.
2 Qualitative and Quantitative Composition
250 mg vial.
Each vial of powder contains 250 mg of dexrazoxane.
500 mg vial.
Each vial of powder contains 500 mg of dexrazoxane.
For the full list of excipients, see Section 6.1.3 Pharmaceutical Form
Dexrazoxane-Reach is an off white to pale yellow lyophilized cake/powder.
When reconstituted, yields a clear, colourless to light pink solution, free of visible particles.
4.1 Therapeutic Indications
Dexrazoxane-Reach is indicated for reducing the incidence and severity of cardiomyopathy associated with doxorubicin administration in women with metastatic breast cancer who have received a cumulative doxorubicin dose of 300 mg/m2 and who will continue to receive doxorubicin therapy to maintain tumour control.
4.2 Dose and Method of Administration
Dexrazoxane-Reach is a cardioprotective agent for use in conjunction with doxorubicin, is a sterile, pyrogen-free lyophilizate intended for intravenous administration.
Dexrazoxane-Reach must only be administered under the supervision of a physician experienced in the use of cytotoxic agents.
Posology.
Administer dexrazoxane for injection via intravenous infusion over 15 minutes. Do not administer via an intravenous push.
The recommended dosage ratio of dexrazoxane for injection to doxorubicin is 10:1 (e.g. 500 mg/m2 dexrazoxane for injection to 50 mg/m2 doxorubicin). Do not administer doxorubicin before dexrazoxane for injection. Administer doxorubicin within 30 minutes after the completion of dexrazoxane for injection infusion.
Method of administration.
Intravenous use.
For use in one patient on one occasion only. Discard any unused portion.
Recommendations for safe handling. Prescribers should refer to national or recognised guidelines on handling cytotoxic agents when using Dexrazoxane-Reach. Reconstitution should only be carried out by trained staff in a cytotoxic designated area. The preparation should not be handled by pregnant staff.
Use of gloves and other protective clothing to prevent skin contact is recommended. Skin reactions have been reported following contact with Dexrazoxane-Reach. If Dexrazoxane-Reach powder or solution comes into contact with the skin or mucosal surfaces, the affected area should immediately be rinsed thoroughly with water.
Preparation for intravenous administration. Reconstitution of Dexrazoxane-Reach.
Reconstitute the vials with sterile water for injection. Reconstitute either with 25 mL for the 250 mg strength vial and 50 mL for the 500 mg strength vial to give a concentration of 10 mg/mL per vial.
Reconstitute either with 12.5 mL for the 250 mg strength vial and 25 mL for the 500 mg strength vial to give a concentration of 20 mg/mL per vial. The vial contents will dissolve within a few minutes with gentle shaking. The resultant solution has a pH of approximately 1.6. This solution should be further diluted before administration to the patient.
Dilution of Dexrazoxane-Reach.
To avoid the risk of thrombophlebitis at the injection site, Dexrazoxane-Reach should be diluted prior to infusion. Dilute the reconstituted solution with lactated Ringer's injection (Hartmann's solution) or 0.16 M sodium lactate (sodium lactate 11.2% should be diluted by a factor of 6 to reach a concentration of 0.16 M) to the required concentration in intravenous infusion bags for intravenous infusion. Such solutions with a higher pH should be used. The final volume is proportional to the number of vials of Dexrazoxane-Reach used and the amount of infusion fluid for dilution.
The use of larger dilution volumes (with a maximum of 100 mL of additional infusion fluid per 25 mL reconstituted Dexrazoxane-Reach) is usually recommended to increase the pH of the solution. Smaller dilution volumes (with a minimum of 25 mL of additional infusion fluid per 25 mL reconstituted Dexrazoxane-Reach) can be used if needed, based on the haemodynamic status of the patient.
Following reconstitution with sterile water for injections, dexrazoxane for injection is stable for 30 minutes at room temperature or if storage is necessary, up to 4 hours from the time of reconstitution when stored under refrigeration at 2° to 8°C. The pH of the resultant solution is 1.0 to 3.0. Discard unused solutions.
The diluted infusion solutions are stable for six hours at room temperature or if storage is necessary, up to 24 hours when stored under refrigeration at 2° to 8°C. The infusion solutions have a pH of 3.5 to 5.5. Discard unused solutions.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Solutions containing a precipitate should be discarded. Dexrazoxane-Reach is normally a clear colourless to light pink solution immediately on reconstitution, but some variability in colour may be observed over time, which does not indicate loss of activity if the product has been stored as recommended. It is however recommended to dispose of the product if the colour immediately on reconstitution is not clear colourless to light pink.
In patients with moderate to severe renal impairment (creatinine clearance < 40 mL/min) the dexrazoxane dose should be reduced by 50% (see Section 4.4).
The dosage ratio should be kept, i.e. if the anthracycline dose is reduced the dexrazoxane dose should be reduced accordingly.4.3 Contraindications
Dexrazoxane-Reach is contraindicated in children aged 0 to 18 years.
Dexrazoxane-Reach is also contraindicated in the following circumstances:
hypersensitivity to dexrazoxane;
breastfeeding (see Section 4.6);
concomitant vaccination with yellow fever vaccine (see Section 4.5).
4.4 Special Warnings and Precautions for Use
Myelosuppression.
Myelosuppressive effects that may be additive to those of chemotherapy were reported with Dexrazoxane-Reach (see Section 4.8). Cell counts at nadir may be lower in patients treated with dexrazoxane. Haematological monitoring is thus necessary. Leucopenia and thrombocytopenia generally reverse quickly upon cessation of treatment with Dexrazoxane-Reach.
At higher doses of chemotherapy, where the Dexrazoxane-Reach dose exceeds 1000 mg/m2, myelosuppression may increase significantly.
Second primary malignancies.
Since dexrazoxane is a cytotoxic agent, with topoisomerase II inhibition activity, combination of dexrazoxane with chemotherapy may lead to an increased risk of second primary malignancy.
Oncology patients have an increased risk of second primary malignancies, regardless of treatment. Patients who have received cancer therapy also have an increased risk of second primary malignancy.
Acute myeloid leukaemia (AML) has been reported uncommonly in adult breast cancer patients post-marketing (see Section 4.8).
In paediatric patients, second primary malignancies, including acute myeloid leukaemia (AML) and myelodysplastic syndrome (MDS), have been reported in clinical trials in both dexrazoxane and control groups. Although second primary malignancies were numerically higher in the dexrazoxane arm, there was no statistical difference between groups. Overall, the rates of second primary malignancies in the available paediatric studies in the dexrazoxane group are similar to rates determined for relevant populations in other studies (historical data). However, the long-term effect of dexrazoxane on second primary malignancies is not known and cannot be estimated from the available data. In clinical trials, second primary malignancies, in particular AML and myelodysplastic syndrome (MDS), have been reported in paediatric patients with Hodgkin's disease and acute lymphoblastic leukaemia receiving chemotherapy regimens including several cytotoxics (e.g. etoposide, doxorubicin, cyclophosphamide) (see Section 4.8).
Patients with cardiac disorders.
Standard cardiac monitoring associated with doxorubicin or epirubicin treatment should be continued.
There are no data that support the use of dexrazoxane in patients with myocardial infarction, preexisting heart failure (including clinical heart failure secondary to anthracycline treatment), uncontrolled angina or symptomatic valvular heart disease.
Thromboembolism.
Combination of dexrazoxane with chemotherapy may lead to an increased risk of thromboembolism (see Section 4.8).
Anaphylactic reaction.
Anaphylactic reaction including angioedema, skin reactions, bronchospasm, respiratory distress, hypotension and loss of consciousness have been observed in patients treated with dexrazoxane and anthracyclines (see Section 4.8). Previous history of allergy to dexrazoxane should be carefully considered prior to administration (see Section 4.3).
Use in hepatic impairment.
Since liver dysfunction was occasionally observed in patients treated with dexrazoxane (see Section 4.8), it is recommended that routine liver function tests be performed before and during administration of dexrazoxane in patients with known liver function disorders.
Use in renal impairment.
Clearance of dexrazoxane and its active metabolites may be reduced in patients with decreased creatinine clearance (see Section 4.2).
Use in elderly.
There are no clinical trials comparing the efficacy or safety of dexrazoxane in geriatric patients to that in younger patients. However, in general, caution is required when treating elderly patients due to their greater use of other medicinal products, higher rates of concomitant diseases and possible reduced hepatic, renal or cardiac function.
Paediatric use.
The safety and efficacy of dexrazoxane in children aged 0 to 18 years have not been established. Currently available data are described in Section 4.3; Section 4.4; Section 4.8; Section 5.1; Section 5.2.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Dexrazoxane-Reach is excreted unchanged via the kidney, as well as metabolized by dihydropyrimidine amidohydrolase (DHPase) in the liver and kidney and dihydroorotase (DHOase) to ring-opened metabolites. Co-administration of doxorubicin (50 to 60 mg/m2) or epirubicin (60 to 100 mg/m2) did not affect Dexrazoxane-Reach pharmacokinetics significantly.
In studies, Dexrazoxane-Reach did not affect the pharmacokinetics of doxorubicin. There is limited evidence from studies that suggests that epirubicin clearance may be increased when dexrazoxane is pre-administered, this occurred at high doses of epirubicin (120-135 mg/m2).
Dexrazoxane-Reach may increase haematological and testicular toxicity induced by chemotherapy or radiation, requiring careful monitoring of haematological parameters during the first two treatment cycles (see Section 4.4).
Dexrazoxane-Reach should not be mixed with any other medicinal products during infusion.
Interference with chemotherapy.
Since both dexrazoxane and anthracyclines are topoisomerase inhibitors, it has been suggested that dexrazoxane may interfere with the anti-tumour efficacy of anthracyclines based on mechanism of action. However, in most adult studies no significant difference has been identified in response rate and overall survival between dexrazoxane and control groups. A significant decrease in tumour response rate was reported in one study of advanced breast cancer patients treated with doxorubicin and dexrazoxane compared to patients treated with doxorubicin and placebo. In this study placebo response rate was considered to be high (60.5%), which may be a contributing factor to the observed difference in response rate. Despite the difference in response rates, there was no significant difference in time to progression or overall survival between patients that had received either dexrazoxane or placebo in this study.
No paediatric study has reported a difference in oncological outcome (event free survival) between groups treated with dexrazoxane and those treated with anthracycline alone.
Concomitant use contraindicated.
Yellow fever vaccine.
Risk of fatal generalised vaccine disease (see Section 4.3).
Concomitant use not recommended.
Other live attenuated vaccines.
Risk of systemic, possible fatal disease. This risk is increased in subjects who are already immunosuppressed by their underlying disease. Use an inactivated vaccine where this exists (poliomyelitis).
Phenytoin.
Cytotoxic agents may reduce the absorption of phenytoin leading to an exacerbation of convulsions. Dexrazoxane is not recommended in combination with phenytoin.
Concomitant use to assess carefully.
Ciclosporin, tacrolimus.
Excessive immunosuppression with risk of lymphoproliferative disease.
Paediatric population.
Interaction studies have only been performed in adults.4.6 Fertility, Pregnancy and Lactation
Women of childbearing potential/contraception in males and females.
Both sexually active men and women should use effective methods of contraception during treatment. For women and men the contraception should be continued for at least 6 months after cessation of treatment with Dexrazoxane-Reach.
Effects on fertility.
The effect of Dexrazoxane-Reach on the fertility of humans has not been studied. There are no fertility data from animal studies available. Testicular atrophy was observed in rats and dogs following repeat dosing at subclinical doses. Dexrazoxane-Reach may impair fertility in males of reproductive potential.
(Category D)
There are no adequate data from the use of dexrazoxane in pregnant women.
Dexrazoxane is embryotoxic and teratogenic in rats and rabbits. Dexrazoxane resulted in maternal toxicity in rats at doses ≥ 2 mg/kg (0.02 times the human dose on a mg/m2 basis) and embryotoxicity and teratogenicity at 8 mg/kg (0.1 times the human dose on a mg/m2 basis) when given daily to pregnant rats during the period of organogenesis. Teratogenic effects in the rat included imperforate anus, microphthalmia, and anophthalmia. In offspring allowed to develop to maturity, fertility was impaired in the male and female rats treated in utero during organogenesis at 8 mg/kg. In rabbits, doses of ≥ 5 mg/kg (0.12 times the human dose on a mg/m2 basis) daily during the period of organogenesis caused maternal toxicity and doses of 20 mg/kg (similar to the human dose on a mg/m2 basis) were embryotoxic and teratogenic. Effects in the rabbit included several skeletal malformations such as short tail, rib and thoracic malformations, and soft tissue variations including subcutaneous, eye and cardiac haemorrhagic areas, as well as agenesis of the gallbladder and of the intermediate lobe of the lung.
The potential risk for humans is unknown. Dexrazoxane-Reach is used with anthracyclines known to have cytotoxic, mutagenic and embryotoxic properties. Dexrazoxane-Reach should not be used during pregnancy unless clearly necessary.
Both sexually active men and women of childbearing potential should be advised not to father a child/become pregnant and must use effective contraceptive measures during and up to 6 months after treatment. Women must inform their doctor immediately if they become pregnant.
There are no animal studies on the transfer of the active substance and/or its metabolites into milk. It is unknown whether dexrazoxane and/or its metabolites are excreted in human milk. Because of the potential for serious adverse reactions in infants exposed to Dexrazoxane-Reach, breastfeeding is contraindicated during Dexrazoxane-Reach treatment (see Section 4.3).4.7 Effects on Ability to Drive and Use Machines
Dexrazoxane-Reach has moderate influence on the ability to drive and use machines. Patients should be advised to be cautious when driving or using machines if they experience fatigue during treatment with Dexrazoxane-Reach.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
Dexrazoxane-Reach is administered together with anthracycline chemotherapy and, consequently, the relative contributions of anthracycline and Dexrazoxane-Reach to the adverse reaction profile may be unclear. The most common adverse reactions are haematological and gastroenterological reactions, primarily anaemia, leukopenia, nausea, vomiting and stomatitis, as well as asthenia and alopecia. Myelosuppressive effects of dexrazoxane may be additive to those of chemotherapy (see Section 4.4).
Tabulated list of adverse reactions.
The following table (see Table 1) includes reactions from clinical trials and from post-marketing use. Due to the spontaneous nature of post-marketing reporting, such events are listed with frequency "not known" if they were not already identified as reactions from clinical trials.
Adverse reactions are ranked under headings of frequency, the most frequent first, using the following convention: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); not known (cannot be estimated from the available data).

Clinical trial data.
Table 1 shows adverse reactions reported in clinical studies and having a reasonable possibility of a causal relationship with dexrazoxane. These data are derived from clinical trials in cancer patients where dexrazoxane was used in combination with anthracycline-based chemotherapy, and where in some cases a control group of patients receiving chemotherapy alone can be referred to.
Patients receiving chemotherapy and dexrazoxane (n=375). Of these 76% were treated for breast cancer and 24% for a variety of advanced cancers.
Dexrazoxane treatment.
A mean dose of 1010 mg/m2 (median: 1000 mg/m2) in combination with doxorubicin, and a mean dose of 941 mg/m2 (median: 997 mg/m2) in combination with epirubicin.
Chemotherapy treatment received by patients treated for breast cancer.
45% combination therapy with doxorubicin 50 mg/m2 (mainly with 5-fluorouracil and cyclophosphamide): 17% with epirubicin alone; 14% combination therapy with epirubicin 60 or 90 mg/m2 (mainly with 5-fluorouracil and cyclophosphamide).
Patients receiving chemotherapy alone (n=157). All were treated for breast cancer.
Chemotherapy treatment received.
43% single agent epirubicin 120 mg/m2; 33% combination therapy with 50 mg/m2 doxorubicin (mainly with 5-fluorouracil and cyclophosphamide); 24% combination therapy with epirubicin at 60 or 90 mg/m2 (mainly with 5-fluorouracil and cyclophosphamide).
Description of selected adverse drug reactions.
Second primary malignancies.
AML has been reported uncommonly in adult breast cancer patients post-marketing.
Safety profile at maximum tolerated dose.
Dexrazoxane's maximum tolerated dose (MTD) when given as monotherapy by short infusion every three weeks for cardioprotection has not been specifically studied. In studies of dexrazoxane as a cytotoxic, its MTD is shown to be dependent on posology and dosing schedule and varies from 3750 mg/m2 when short infusions are given in divided doses over 3 days to 7420 mg/m2 when given weekly for 4 weeks, with myelosuppression and abnormal liver function tests becoming dose-limiting. The MTD is lower in patients who have been heavily pre-treated with chemotherapy, and those with pre-existing immunosuppression (e.g. AIDS).
The following are adverse reactions reported when dexrazoxane was given at doses around the MTD: neutropenia, thrombocytopenia, nausea, vomiting, and increase in hepatic parameters. Other toxic effects were malaise, low grade fever, increased urinary clearance of iron and zinc, anaemia, abnormal blood clotting, transient elevation of serum triglyceride and amylase levels, and a transient decrease in serum calcium level.
Paediatric population.
The safety experience in children is based primarily on literature reports of clinical trials in acute lymphoblastic leukaemia, non-Hodgkin's lymphoma, Hodgkin's disease and osteosarcoma, and post-marketing data.
In paediatric patients, second primary malignancies, including acute myeloid leukaemia (AML) and myelodysplastic syndrome (MDS), have been reported in clinical trials in both dexrazoxane and control groups. Although second primary malignancies were numerically higher in the dexrazoxane arms, there was with no statistical difference between groups. In addition, the long-term effect of dexrazoxane on secondary primary malignancies is not known (cannot be estimated from the available data) (see Section 4.4).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
The signs and symptoms of overdose are likely to consist of leucopenia, thrombocytopenia, nausea, vomiting, diarrhoea, skin reactions and alopecia. There is no specific antidote and symptomatic treatment should be provided.
Management should include prophylaxis and treatment of infections, fluid regulation, and maintenance of nutrition.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: detoxifying agents for antineoplastic treatment, ATC code: V03AF02.
Mechanism of action.
The exact mechanism by which dexrazoxane exerts its cardioprotective effect has not been fully elucidated, however based on the available evidence the following mechanism has been suggested. The dose-dependent cardiotoxicity observed during anthracycline administration is due to anthracycline-induced iron-dependent free radical oxidative stress on the relatively unprotected cardiac muscle. Dexrazoxane, an analogue of EDTA (ethylene diamine tetra-acetic acid), is hydrolysed in cardiac cells to the ring-opened product ICRF-198. ICRF-198 is capable of chelating metal ions. It is generally thought that they can provide cardioprotection by scavenging metal ions thus preventing the Fe3+-anthracycline complex from redox cycling and forming reactive radicals. Dexrazoxane also inhibits topoisomerase IIb, which may also contribute to protection of anthracycline-induced cardiotoxicity.
Clinical trials.
The evidence from clinical trials to date suggests increasing cardioprotective benefit from dexrazoxane as the cumulative anthracycline dose is increased.
Dexrazoxane does not protect against non-cardiac toxicities induced by anthracyclines.
The majority of controlled clinical studies were performed in patients with advanced breast cancer and employed a dosing ratio of dexrazoxane: doxorubicin of 20:1 or 10:1. In two clinical studies that used the higher dose ratio (one in breast cancer and one in small cell lung cancer) a higher rate of death was reported in the groups treated with dexrazoxane plus chemotherapy compared to those treated with chemotherapy alone or with placebo. The dose ratio was subsequently reduced to 10:1 in both studies, and no significant differences in survival were reported in patients treated at the lower dose ratio. However, a number of studies that used the higher dose ratio throughout have not reported any difference in survival.
Paediatric population.
There are limited data on efficacy in children. Data are mainly derived from COG (Children's Oncology Group) studies, published in BL Asselin et al: J. Clin. Oncol. 2016 and CL Schwartz et al Pediatr. Blood Cancer 2016.
Study P9404 (BL Asselin et al: J. Clin. Oncol. 2016) evaluated the cardioprotective efficacy, and safety of dexrazoxane added to chemotherapy that included a cumulative doxorubicin dose of 360 mg/m2 to treat children and adolescents with newly diagnosed T-cell acute lymphoblastic leukemia (T-ALL) or lymphoblastic non-Hodgkin lymphoma (L-NHL). Between June 1996 and September 2001, patients were randomised to receive doxorubicin treatment with (n=273) or without (n=264) dexrazoxane (dexrazoxane: doxorubicin ratio 10:1). Dexrazoxane was given as a bolus infusion immediately before every dose of doxorubicin. Cardiac effects were assessed by echocardiographic measurements of left ventricular function and structure.
Baseline characteristics of the overall study population were as follows: median age at diagnosis 9.2 years, male (75.8%), White (66%), T-ALL (67%). The treatment used was modified from study protocol DFCI ALL-87-01 with or without high-dose methotrexate and all patients received cranial radiation.
Heart failure was not reported among patients at any time during treatment or follow-up. Of the five patients in whom grade 3 or 4 cardiac toxicity occurred while receiving therapy; two had arrhythmias (n = 1 in the dexrazoxane group), and three had decreased LV fractional shortening (all were in the no-dexrazoxane group). All five received high-dose methotrexate and had a serious infection when the cardiac toxicity occurred. All patients recovered and completed chemotherapy, including doxorubicin. cTnT levels at both baseline and during treatment were available for 160 patients. The probability of having elevated cTnT was lower in the dexrazoxane group (odds ratio, 0.23; 95% CI, 0.05 to 1.11; p = 0.067).
At baseline, mean z scores for LV fractional shortening and LV thickness-to-dimension ratio were similar between treatment groups. The mean z score for LV wall thickness at baseline in the dexrazoxane-treated group was significantly lower than in the no-dexrazoxane group. The LV wall thickness was worse after treatment in the no-dexrazoxane treatment group than it was in the dexrazoxane-treated group. After doxorubicin treatment, mean z scores were lower than age expected norms for all children, but were not significantly different between groups; the mean score was always closer to normal for the dexrazoxane group. The mean z scores for left ventricular fractional shortening, LV wall thickness, and LV thickness-to-dimension ratio at three years in dexrazoxane treated children were not significantly different from the scores for healthy children; whereas in the no-dexrazoxane group these z scores all remained significantly reduced compared to healthy children. The mean left ventricular fractional shortening, wall thickness, and thickness-to-dimension ratio z scores measured 3 years after diagnosis were worse in the doxorubicin-alone group (n = 55 per group; P ≤ 0.01 for all comparisons).
The 5-year event-free survival (with standard error) did not differ between groups: 76.7% (2.7%) for the dexrazoxane group versus 76.0% (2.7%) for the doxorubicin-only group (p = 0.9) (also see Section 4.2; Section 4.3; Section 4.4; Section 4.8). The frequencies of severe grade 3 or 4 haematologic toxicity, infection, central nervous system events, and toxic deaths were similar in both groups.
In a non-randomised study (P9754, CL Schwartz et al Pediatr. Blood Cancer 2016) in patients with non-metastatic osteosarcoma (median age 13 years, range 3-30 years) where all patients receiving doxorubicin (450-600 mg/m2) also received dexrazoxane (dexrazoxane: doxorubicin ratio 10:1) (242 patients exposed to at least 450 mg/m2 doxorubicin and 101 exposed to 600 mg/m2), grade 1 or 2 left ventricular dysfunction occurred in five patients, and was transient in at least four of these. In two of these patients, doxorubicin was subsequently discontinued. No grade 3, 4 or 5 cardiomyopathy (ventricular dysfunction) was observed. One additional patient had grade 3 serum cTnT elevation with 600 mg/m2 doxorubicin without documented myocardial dysfunction. Left ventricular fractional shortening values from 104 evaluable patients were converted to z-scores (FSZ) to review change in cardiac function since time of enrolment. It was found that FSZ decreased in a statistically significant fashion with increasing time, with that change being -0.017 ± 0.009 of a standardised unit (z-score of 1) per week (estimated annual change of 0.9 FSZ units). Assignment to standard therapy (450 mg/m2 doxorubicin) or intensification (600 mg/m2 doxorubicin) was unrelated to change in FSZ. In terms of clinical cardiotoxicity, biomarker measures and FSZ analysis, the risk of acute cardiomyopathy was low, given the cumulative doses of 450 mg/m2 to 600 mg/m2 of doxorubicin (also see Section 4.2; Section 4.3; Section 4.4; Section 4.8).
5.2 Pharmacokinetic Properties
After intravenous administration to cancer patients, serum kinetics of dexrazoxane generally follow an open two-compartment model with first-order elimination. The maximum plasma concentration observed after a 12-15-minute infusion of 1000 mg/m2 is around 80 microgram/mL with area under the plasma concentration-time curve (AUC) of 130 ± 27 mg.h/L. The plasma concentrations declined thereafter with an average half-life value of 2.2 ± 0.42 hours. The total body clearance of dexrazoxane in adults is estimated at 14.4 ± 2.8 L/h.
Distribution.
The apparent volume of distribution is 44.0 ± 3.9 L, suggesting that dexrazoxane distributes mainly in the total body water. Plasma protein binding of dexrazoxane is low (2%) and it does not penetrate into the cerebrospinal fluid to a clinically significant extent.
Dexrazoxane and its metabolites were detected in the plasma and urine of animals and man.
Elimination.
Urinary excretion plays an important role in the elimination of dexrazoxane. The total urinary excretion of unchanged dexrazoxane is in the order of 40%.
Special populations.
Paediatric patients.
The very limited pharmacokinetic data in children suggests that although absolute values of clearance are higher, values normalised for body surface area are not significantly different from those of adults.
Geriatric patients.
No studies have been conducted in the elderly and dexrazoxane. Clearance may be reduced in elderly patients and patients with low creatinine clearance.
Hepatic impairment.
No studies have been conducted in subjects with hepatic impairment.
Renal impairment.
Compared with normal subjects (creatinine clearance (ClCr) > 80 mL/min), exposure was 2-fold greater in subjects with moderate (ClCr of 30 to 50 mL/min) to severe (ClCr < 30 mL/min) renal impairment. Modelling suggested that equivalent exposure (AUC0-inf) could be achieved if dosing were reduced by 50% in subjects with ClCr less than 40 mL/min compared with control subjects (ClCr > 80 mL/min).
5.3 Preclinical Safety Data
Genotoxicity.
Dexrazoxane was not mutagenic in the Ames bacterial assay. Dexrazoxane was clastogenic in human lymphocytes and human lymphoblastoid cells in vitro. Dexrazoxane and/or razoxane (the racemic mixture of which dexrazoxane is the S (+)-enantiomer) was also clastogenic in mice and Chinese hamsters in bone marrow cell micronucleus and chromosome aberration assays. Thus, dexrazoxane is genotoxic.
Carcinogenicity.
The carcinogenic potential of dexrazoxane has not been investigated. However prolonged administration of high doses of razoxane, the racemic mixture of which dexrazoxane is the S (+) enantiomer, has been associated with the development of hematopoietic neoplasms in female mice, lymphocytic neoplasms in female mice and uterine adenocarcinomas in female rats.6 Pharmaceutical Particulars
6.1 List of Excipients
Hydrochloric acid.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Before opening: store below 25°C. In order to protect from light store in the original package.
Use as soon as practicable. After reconstitution, store no more than 4 hours at 2-8°C. Contains no antimicrobial preservative.
To reduce microbiological hazard, use as soon as practicable after dilution. If storage is necessary, hold for 6 hours at room temperature or at 2-8°C for not more than 24 hours.
6.5 Nature and Contents of Container
Each 250 mg vial contains 250 mg of powder.
The 250 mg vial is a 20 mm neck 30 mL clear moulded glass vial with 20 mm grey bromobutyl Igloo rubber stopper and sealed with 20 mm aluminium flip of seal.
Each 500 mg vial contains 500 mg of powder.
The 500 mg is a 20 mm neck 50 mL clear moulded glass vial with 20 mm grey bromobutyl Igloo rubber stopper and sealed with 20 mm aluminium flip of seal.
Dexrazoxane-Reach 250 mg is supplied in packs of 1 vial.
Dexrazoxane-Reach 500 mg is supplied in packs of 1 vial.
6.6 Special Precautions for Disposal
Disposal.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements. Adequate care and precautions should be taken in the disposal of items used to reconstitute and dilute Dexrazoxane-Reach.6.7 Physicochemical Properties
Chemically, dexrazoxane is (S)-4,4'-(1-methyl-1,2-ethanediyl) bis-2,6-piperazinedione.
Chemical structure.
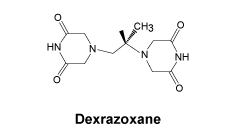 It has the molecular formula C11H16N4O4 and a molecular weight of 268.28.
It has the molecular formula C11H16N4O4 and a molecular weight of 268.28.7 Medicine Schedule (Poisons Standard)
S4 - Prescription only medicine.
Summary Table of Changes
