What is in this leaflet
This leaflet answers some common questions about DILART.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine. All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What DILART is used for
DILART belongs to a group of medicines called angiotensin II receptor antagonists (AIIRAs).
Hypertension
DILART is used to control high blood pressure, also called hypertension. Everyone has blood pressure. This pressure helps get your blood around your body. Your blood pressure may be different at different times of the day, depending on how busy or worried you are. You have hypertension when your blood pressure stays higher than is needed, even when you are calm and relaxed.
There are usually no symptoms of hypertension. The only way of knowing that you have it is to have your blood pressure checked regularly.
High blood pressure increases the workload of the heart and blood vessels. If it continues for a long time, it can damage the blood vessels in the brain, heart and kidneys. This can lead to stroke, heart failure or kidney failure. High blood pressure increases the risk of heart attacks. Lowering your blood pressure reduces the chance of these disorders happening.
Heart Failure
DILART is used to treat heart failure.
Heart failure means that the heart muscle cannot pump blood strongly enough to supply all the blood needed throughout the body. Heart failure is not the same as heart attack and does not mean that the heart stops. DILART helps the heart to function better and relieves some of the symptoms of heart failure.
Heart Attack
DILART is also used to treat people after they have had a heart attack (myocardial infarction) to reduce the risk of further heart damage and reduce further heart problems.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another purpose.
There is not enough information to recommend the use of DILART in children (below 18 years of age).
This medicine is only available with a doctor's prescription. It is not habit-forming.
Before you take DILART
When you must not take it
Do not take DILART if you have ever had an allergic reaction after taking:
- valsartan (the active ingredient in DILART)
- any of the other ingredients listed at the end of this leaflet
Symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
Do not take DILART if you are pregnant or intend to become pregnant. DILART is not recommended for use in pregnancy. Like other similar medicines, it could affect your unborn baby.
Do not take DILART if you have liver disease caused by a blockage in the bile duct or any other severe liver disease. DILART is not recommended if you have these conditions.
Do not take DILART if you are also taking other blood pressure lowering medicines containing aliskiren and have type 2 diabetes.
Do not take DILART after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. In that case, return it to your pharmacist for disposal.
Before you start to take it
Tell your doctor if you have any of the following health problems/medical conditions:
- heart disease or high blood pressure that is being treated with large doses of diuretics (also called water or fluid tablets), or being treated with beta-blockers, aliskiren and/or ACE-inhibitors
- high blood pressure due to narrowing of the arteries in the kidney
- any other kidney problems or are having dialysis
- milder forms of liver disease
- swelling, mainly of the face and throat, while taking other medicines (including an ACE inhibitor or aliskiren)
- you have recently had severe vomiting or diarrhoea
- you are severely limiting your salt intake
- primary hyperaldosteronism (Conn's syndrome), a hormone disorder causing fluid retention
- obstructed blood flow through the heart from narrowing of valves (stenosis) or enlarged septum of the heart (HOCM)
Your doctor may want to take special precautions if you have any of the above conditions.
Tell your doctor if you are breast-feeding. Ask your doctor about the risks and benefits of taking DILART in this case. It is not known if valsartan, the active ingredient of DILART, passes into the breast milk and could affect your baby.
Tell your doctor if you are allergic to any other medicines, foods, dyes or preservatives. Your doctor will want to know if you are prone to allergies.
If you have not told your doctor about any of these things, tell them before you take DILART.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including medicines that you buy without a prescription from a pharmacy, supermarket or health food shop.
Other medicines may be affected by DILART or they may affect how well DILART works. You may need to take different amounts of your medicines or you may need to take different medicines.
These medicines include:
- beta-blockers, which are medicines used to treat hypertension or other heart conditions
- ACE-inhibitors or aliskiren, which are also medicines used to treat hypertension or other heart conditions
- some diuretics (water or fluid pills)
- potassium supplements (e.g. Slow-K®) or other drugs that may increase potassium levels
- salt substitutes containing potassium
- lithium (a medicine used to treat some types of psychiatric illness)
- some antibiotics (rifamycins), anti-rejection drugs (ciclosporin), antiretrovirals (ritonavir) which may increase the effect of DILART
- anti-inflammatory medicines such as Celebrex, Voltaren and Indocid (NSAIDs) or Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors).
- trimethoprim containing medicines
Your doctor may also check your kidney function.
Your doctor and pharmacist have a more complete list of medicines to be careful of while taking DILART.
How to take DILART
Follow carefully all directions given to you by your doctor and pharmacist. These instructions may differ from the information contained in this leaflet.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
For hypertension, the usual dose is one 80 mg tablet once a day. If your blood pressure is still too high after 4 weeks, your doctor may increase the dose to 160 mg once a day, or from 160 mg to 320 mg once a day. If your blood pressure is still too high, your doctor may add a different type of blood pressure lowering medicine.
For heart failure the usual starting dose is 40 mg twice daily. Your doctor may increase the dose gradually up to one 160 mg tablet twice daily.
Following a heart attack, treatment is generally started at a dose of 20 mg (half a 40 mg tablet) twice daily. Your doctor may increase the dose gradually up to 160 mg twice daily.
When to take it
When you take the first tablet from the pack of DILART, take the one marked with the correct day of the week (e.g. if it is Wednesday, take the tablet marked Wednesday). DILART comes in a calendar pack with the days of the week marked on it to help you remember to take your tablet each day.
Take it at the same time each day. This also helps you remember to take it, especially if you take it as part of your usual routine (e.g. at breakfast time).
How to take it
Swallow the tablet with a full glass of water. Always take it in the same way in relation to food. It does not matter if you take it after food or on an empty stomach, as long as you take it the same way each day.
If your stomach is upset after taking DILART, always take it after a meal (e.g. breakfast).
How long to take it
Take this medicine until your doctor tells you to stop even if you feel quite well. It will take at least 4 weeks for this medicine to have its full effect. After that, it will be continued for as long as your doctor thinks is needed.
If you forget to take it
If it is almost time for your next dose, skip the missed dose and take the next one when you are meant to. Otherwise, take the dose as soon as you remember and then go back to taking it as you would normally.
Do not take a double dose to make up for the one that you missed. This may increase the chance of you getting unwanted side effects.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone number: 131126) for advice, or go to Accident and Emergency at your nearest hospital if you think that you or anyone else may have taken too much DILART. Do this even if there are no signs of discomfort or poisoning.
Keep the telephone numbers for these places handy.
Too much DILART may make you feel dizzy, lightheaded or faint. You may experience rapid, shallow breathing or cold, clammy skin. Your heartbeat may be faster than usual. This is because your blood pressure is too low.
While you are taking DILART
Things you must do
If you become pregnant while taking DILART, tell your doctor immediately. You should not take this medicine while you are pregnant.
Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed. Otherwise your doctor may think that it was not effective and change your treatment unnecessarily.
Be sure to keep all of your doctor's appointments so that your progress can be checked. Do this even if you feel well. It is important to keep track of your progress. Your doctor will want to check your blood pressure and your kidney and liver function from time to time.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking DILART.
Tell any other doctor, dentist or pharmacist who treats you that you are taking DILART.
Things you must not do
Do not use DILART to treat any other complaints unless your doctor says you can.
Do not give this medicine to anyone else, even if their condition seems to be similar to yours.
Things to be careful of
Be careful driving, operating machinery or doing jobs that require you to be alert while you are taking DILART until you know how it affects you. This medicine can cause tiredness, sleepiness or dizziness in some people. If you have these symptoms, do not drive or do anything else that could be dangerous.
If this medicine makes you feel dizzy or light-headed, be careful when getting up from a sitting or lying position. Dizziness can usually be prevented by getting up slowly and flexing leg muscles and toes to get the blood flowing. When getting out of bed, dangle your legs over the side for a minute or two before standing up.
Lifestyle measures that help reduce heart disease risk
By following these simple measures, you can further reduce the risk from heart disease.
- Quit smoking and avoid second-hand smoke.
- Limit alcohol intake.
- Enjoy healthy eating by:
- eating plenty of vegetables and fruit;
- reducing your saturated fat intake (eat less fatty meats, full fat dairy products, butter, coconut and palm oils, most take-away foods, commercially-baked products). - Be active. Progress, over time, to at least 30 minutes of moderate-intensity physical activity on 5 or more days each week. Can be accumulated in shorter bouts of 10 minutes duration. If you have been prescribed anti-angina medicine, carry it with you when being physically active.
- Maintain a healthy weight.
- Discuss your lifestyle and lifestyle plans with your doctor.
- For more information and tools to improve your heart health, call Heartline, the Heart Foundation's national telephone information service, on 1300 36 27 87 (local call cost).
Know warning signs of heart attack and what to do:
- Tightness, fullness, pressure, squeezing, heaviness or pain in your chest, neck, jaw, throat, shoulders, arms or back.
- You may also have difficulty breathing, or have a cold sweat or feel dizzy or light headed or feel like vomiting (or actually vomit).
- If you have heart attack warning signs that are severe, get worse or last for 10 minutes even if they are mild, call triple zero (000). Every minute counts.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking DILART, even if you do not think it is connected with the medicine. All medicines can have side effects. Sometimes they are serious, but most of the time they are not. You may need medical treatment if you get some of the side effects.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of these side effects and they worry you:
- headache
- dizziness, spinning sensation (vertigo)
- sleepiness, tiredness or weakness
- diarrhoea, constipation or wind
- nausea (feeling sick), vomiting, loss of appetite, stomach pains or indigestion
- dry cough, sore throat or hoarse voice
- runny nose or congested sinuses
- pain in the back or joints
- muscle pain or cramps
- difficulty sleeping
- feeling anxious
- tingling or numbness in the hands or feet
- problems with sexual function
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- signs of allergy such as rash, itching or hives on the skin; swelling of the face, lips, tongue or other part of the body; fever, shortness of breath, wheezing or troubled breathing
- feeling of fast or irregular heart beat (pounding, racing, skipping beats)
- chest pain
- shortness of breath not caused by exercise, with swelling of legs or feet
- tiredness or lack of energy, being short of breath when exercising, dizziness and looking pale
- constant "flu-like" symptoms such as chills, fever, sore throat, aching joints, sores in mouth, swollen glands
- severe dizziness or fainting
- liver disease with nausea, vomiting, loss of appetite, feeling generally unwell, fever, itching, yellowing of the skin and eyes, and dark coloured urine
The above side effects may be serious. You may need urgent medical attention.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some people.
After using DILART
Storage
Keep your tablets in the original container until it is time to take them.
Store them in a cool dry place (room temperature).
Do not store DILART or any other medicine in the bathroom or near a sink.
Do not leave it in the car or on window sills. Heat and dampness can destroy some medicines. DILART will keep well if it is cool and dry.
Keep the medicines where children cannot reach them. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking DILART, or the tablets have passed their expiry date, ask your pharmacist what to do with any that are left over.
Product description
What it looks like
DILART tablets are supplied in blister packs of 28.
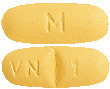
DILART 40 mg tablets are yellow, oval shaped, biconvex, film coated tablets debossed with "VN" and "1" on either side of the score line on one side and "M" on the other side.
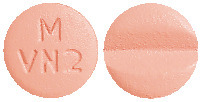
DILART 80 mg tablets are pale red, round, biconvex, bevelled edge, film-coated tablets having a score line on one side and debossed with "M" over "VN 2" on other side.
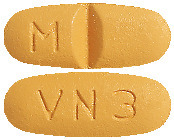
DILART 160 mg tablets are beige, oval-shaped, biconvex, bevelled edge, film-coated tablets debossed with "M" to the left of the score on one side and "VN 3" on the other side.
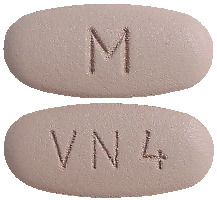
DILART 320 mg tablets are dark grey, oval shaped, biconvex, film coated tablets debossed with "VN 4" on one side and "M" on the other side.
Ingredients
The tablets contain the following non active ingredients:
- microcrystalline cellulose
- crospovidone
- povidone
- croscarmellose sodium
- colloidal anhydrous silica
- magnesium stearate
- The 40 mg tablet contains:
Opadry Complete Film Coating System 08F520002 Yellow (ARTG PI No: 144620) - The 80 mg tablet contains:
Opadry Complete Film Coating System 08F540003 Pink (ARTG PI No: 146273) - The 160 mg tablet contains
Opadry Complete Film Coating System 08F520000 Yellow (ARTG PI No: 146272) - The 320 mg tablet contains:
Opadry Complete Film Coating System 08F565001 Brown (ARTG PI No: 146276)
Sponsor
DILART is supplied in Australia by:
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
Phone: 1800 274 276
www.viatris.com.au
This leaflet was prepared in November 2023.
Australian registration numbers:
DILART 40 mg (blister) –
AUST R 167425
DILART 80 mg (blister) –
AUST R 167427
DILART 160 mg (blister) –
AUST R 167426
DILART 320 mg (blister) –
AUST R 167421
DILART ® is a Viatris company trade mark
DILART_cmi\Nov23/00
Published by MIMS January 2024

 Other adverse experiences that occurred in controlled clinical trials of patients treated with valsartan (> 0.2% of valsartan patients) are listed below. It cannot be determined whether these events were causally related to valsartan.
Other adverse experiences that occurred in controlled clinical trials of patients treated with valsartan (> 0.2% of valsartan patients) are listed below. It cannot be determined whether these events were causally related to valsartan.

 Since this was a trial with an active control (captopril), an additional analysis of all cause mortality was performed to estimate how valsartan would have performed versus placebo. Using the results of the previous reference myocardial infarction trials - SAVE, AIRE, and TRACE - the estimated effect of valsartan preserved 99.6% of the effect of captopril (97.5% CI = 60-139%). Combining valsartan with captopril did not add further benefit over captopril alone, therefore this combination is not recommended. There was no difference in all-cause mortality based on age, gender, race, baseline therapies or underlying disease.
Since this was a trial with an active control (captopril), an additional analysis of all cause mortality was performed to estimate how valsartan would have performed versus placebo. Using the results of the previous reference myocardial infarction trials - SAVE, AIRE, and TRACE - the estimated effect of valsartan preserved 99.6% of the effect of captopril (97.5% CI = 60-139%). Combining valsartan with captopril did not add further benefit over captopril alone, therefore this combination is not recommended. There was no difference in all-cause mortality based on age, gender, race, baseline therapies or underlying disease. Molecular formula: C24H29N5O3. Molecular weight: 435.5.
Molecular formula: C24H29N5O3. Molecular weight: 435.5.