1 Name of Medicine
Avatrombopag maleate.
2 Qualitative and Quantitative Composition
Each Doptelet tablet contains 23.6 mg avatrombopag maleate (equivalent to 20 mg of avatrombopag).
Excipients with known effect.
Sugars as lactose.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Film coated tablet.
Pale yellow, round biconvex, film-coated tablet, debossed with ''AVA'' on one side and ''20'' on the other side.
4.1 Therapeutic Indications
Doptelet is indicated for the treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure.
Doptelet is indicated for the treatment of thrombocytopenia in adult patients with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment.
4.3 Contraindications
Known hypersensitivity to avatrombopag or to any of the inactive ingredients (see Section 6.1 List of Excipients).
4.4 Special Warnings and Precautions for Use
Thrombotic/thromboembolic events.
Doptelet is a thrombopoietin (TPO) receptor agonist and TPO receptor agonists have been associated with thrombotic and thromboembolic complications in patients with chronic liver disease or chronic immune thrombocytopenia. In patients with chronic liver disease, thromboembolic events (portal vein thrombosis) occurred in 0.4% (1/274) of patients receiving Doptelet. In patients with chronic immune thrombocytopenia, thromboembolic events (arterial or venous) occurred in 7% (9/128) of patients receiving Doptelet.
Consider the potential increased thrombotic risk when administering Doptelet to patients with known risk factors for thromboembolism, including genetic prothrombotic conditions (e.g. Factor V Leiden, Prothrombin 20210A, Antithrombin deficiency or Protein C or S deficiency), acquired risk factors (e.g. antiphospholipid syndrome).
Doptelet should not be administered to patients with chronic liver disease or chronic immune thrombocytopenia in an attempt to normalise platelet counts. Follow the dosing guidelines to achieve target platelet counts. Monitor patients receiving Doptelet for signs and symptoms of thromboembolic events and institute treatment promptly.
Use in patients with chronic liver disease undergoing invasive procedures.
The objective of treatment with Doptelet is to increase platelet counts. Doptelet is not expected to correct coagulation factor deficiencies.
The Doptelet clinical studies did not include major surgeries such as laparotomy, thoracotomy, open-heart surgery, craniotomy or excision of organs (see Section 5.1 Pharmacodynamic Properties, Clinical trials, Patients with chronic liver disease). The safety and efficacy of Doptelet has not been established for these procedures.
Reoccurrence of thrombocytopenia and bleeding after cessation of treatment in patients with chronic immune thrombocytopenia.
Thrombocytopenia is likely to reoccur in ITP patients upon discontinuation of treatment with avatrombopag. Following discontinuation of avatrombopag, platelet counts return to baseline levels within 2 weeks in the majority of patients, which increases the bleeding risk and in some cases may lead to bleeding. There is an increased risk of bleeding if avatrombopag treatment is discontinued in the presence of anticoagulants or anti-platelet agents. Patients should be closely monitored for a decrease in platelet count and medically managed to avoid bleeding upon discontinuation of treatment with avatrombopag. It is recommended that, if treatment with avatrombopag is discontinued, ITP treatment be restarted according to current treatment guidelines. Additional medical management may include cessation of anticoagulant and/or antiplatelet therapy, reversal of anticoagulation, or platelet support.
Increased bone marrow reticulin.
Increased bone marrow reticulin is believed to be a result of TPO receptor stimulation, leading to an increased number of megakaryocytes in the bone marrow, which may subsequently release cytokines. Increased reticulin may be suggested by morphological changes in the peripheral blood cells and can be detected through bone marrow biopsy. Therefore, examinations for cellular morphological abnormalities using peripheral blood smear and complete blood count (CBC) prior to and during treatment with avatrombopag are recommended.
If a loss of efficacy and abnormal peripheral blood smear are observed in patients, administration of avatrombopag should be discontinued, a physical examination should be performed, and a bone marrow biopsy with appropriate staining for reticulin should be considered. If available, comparison to a prior bone marrow biopsy should be made. If efficacy is maintained and abnormal peripheral blood smear is observed in patients, the physician should follow appropriate clinical judgment, including consideration of a bone marrow biopsy, and the risk-benefit of avatrombopag and alternative ITP treatment options should be re-assessed.
Progression of existing myelodysplastic syndrome (MDS).
The effectiveness and safety of Doptelet have not been established for the treatment of thrombocytopenia due to MDS. Doptelet should not be used outside of clinical studies for the treatment of thrombocytopenia due to MDS.
There is a theoretical concern that TPO-R agonists may stimulate the progression of existing haematological malignancies such as MDS. TPO-R agonists are growth factors that lead to thrombopoietic progenitor cell expansion, differentiation and platelet production. The TPO-R is predominantly expressed on the surface of cells of the myeloid lineage. For TPO-R agonists there is a concern that they may stimulate the progression of existing haematopoietic malignancies such as MDS.
The diagnosis of ITP in adults and elderly patients should have been confirmed by the exclusion of other clinical entities presenting with thrombocytopenia, in particular the diagnosis of MDS must be excluded. Consideration should be given to performing a bone marrow aspirate and biopsy over the course of the disease and treatment, particularly in patients over 60 years of age, for those with systemic symptoms or abnormal signs such as increased peripheral blast cells.
Co-administration with interferon preparations.
Interferon preparations have been known to reduce platelet counts, therefore, this should be considered when co-administering avatrombopag with interferon preparations.
Use in hepatic impairment.
There is limited information on the use of avatrombopag in patients with severe (Child-Pugh class C, MELD score > 24) hepatic impairment. Avatrombopag should only be used in such patients if the expected benefit outweighs the expected risks (see Section 5.2 Pharmacokinetic Properties).
Patients with severe hepatic impairment should be supported in line with clinical practice by close monitoring for early signs of worsening or new onset hepatic encephalopathy, ascites, and thrombotic or bleeding tendency, through monitoring of liver function tests, tests used for assessing clotting status and through imaging of portal vasculature as needed.
Patients with Child-Pugh class C liver disease who take avatrombopag prior to an invasive procedure, should be evaluated on the day of the procedure for an unexpectedly high increase in platelet count.
Use in the elderly.
Clinical studies of Doptelet did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
Paediatric use.
Safety and efficacy in children have not been established (see Section 5.3 Preclinical Safety Data).
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Patients with chronic liver disease.
No dosage adjustments are required for patients with chronic liver disease.
Effect of other drugs on Doptelet in patients with chronic immune thrombocytopenia.
Moderate or strong dual inhibitors of CYP2C9 and CYP3A4.
Concomitant use with a moderate or strong dual inhibitor of CYP2C9 and CYP3A4 (e.g. fluconazole) increases avatrombopag AUC (see Section 5.2 Pharmacokinetic Properties), which may increase the risk of Doptelet toxicities. Reduce the starting dosage of Doptelet when used concomitantly with a moderate or strong dual inhibitor of CYP2C9 and CYP3A4 (see Table 4) (see Section 4.2 Dose and Method of Administration).
In patients starting moderate or strong dual inhibitors of CYP2C9 and CYP3A4 while receiving Doptelet, monitor platelet counts and adjust Doptelet dose as necessary (see Table 3) (see Section 4.2 Dose and Method of Administration).
Moderate or strong dual inducers of CYP2C9 and CYP3A4.
Concomitant use with a moderate or strong dual inducer of CYP2C9 and CYP3A4 (e.g. rifampicin) decreases avatrombopag AUC (see Section 4.2 Dose and Method of Administration) which may reduce Doptelet efficacy. Increase the recommended starting dosage of Doptelet when used concomitantly with a moderate or strong dual inducer of CYP2C9 and CYP3A4 (see Table 4) (see Section 4.2 Dose and Method of Administration).
In patients starting moderate or strong dual inducers of CYP2C9 and CYP3A4 while receiving Doptelet, monitor platelet counts and adjust dose as necessary (see Table 3) (see Section 4.2 Dose and Method of Administration).
In vitro studies where drug interaction potential was not further evaluated clinically.
In vitro cytochrome P450 (CYP) inhibition and induction studies indicated that avatrombopag does not inhibit CYP 1A, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 or 3A, nor induce CYP 1A, 2B6, 2C or 3A at clinically relevant concentrations. Avatrombopag weakly induces CYP2C8 and CYP2C9. In vitro, avatrombopag inhibits organic anion transporter (OAT)1, OAT3, organic cation transporter (OCT)1, breast cancer resistance protein (BCRP), MRP2, organic anion transporter polypeptide (OATP) 1B3 and P-gp (intestinal), but is not an inhibitor of OATP1B1, bile salt export pump (BSEP), multidrug and toxin compound extrusion protein (MATE)1/2K or OCT2 at clinically meaningful concentrations.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
The effect of avatrombopag on human fertility has not been established, and a risk cannot be ruled out. Fertility was unaffected in male rats treated orally with 100 mg/kg/day avatrombopag (15 times the exposure at the clinical dose for ITP) or in female rats at oral doses of 1000 mg/kg/day (79 times the exposure at the clinical dose for ITP). However, due to differences in TPO receptor specificity, data from nonclinical species do not fully model effects in humans.
(Category B31)
There are no adequate or well-controlled studies with avatrombopag in pregnant women. The effect of Doptelet on human pregnancy is unknown. Doptelet should not be used during pregnancy unless the expected benefit clearly outweighs the potential risk to the fetus.
In embryofetal development studies in rats, an increased incidence of fetal skeletal variations was observed at doses of 300 mg/kg/day and decreased fetal weight were observed at ≥ 1000 mg/kg/day. Spontaneous abortions were observed in rabbits at doses of 100 mg/kg/day. There were no embryofetal effects in rabbits at doses up to 600 mg/kg/day. Findings in both rats and rabbits were associated with maternal toxicity. Plasma exposures (AUC) at the no observed adverse effect level (100 mg/kg/day in rats and 600 mg/kg/day in rabbits) were approximately 37 and 24 times those in humans at the maximum recommended human dose (MRHD) in rats and rabbits, respectively.
In pre-and postnatal development studies in pregnant rats, avatrombopag was administered during both the organogenesis and lactation periods where maternal toxicity leading to total litter loss, decreased body weight in pups, and increased pup mortality was observed at ≥ 100 mg/kg/day. At a dose of 50 mg/kg/day, decreased body weight gain was observed in pups, resulting in a delay in sexual maturation and increased pup mortality from postnatal days 4 to 21, which continued until postnatal day 25. There were no effects on behavioural or reproductive function in the offspring. The effects in pups at 50 mg/kg/day were observed in the absence of maternal toxicity. Plasma exposures (AUC) at the no observed adverse level (15 mg/kg/day) were approximately 9 times the those in humans at the MRHD.
1Australian pregnancy category B3: Drugs which have been taken by only a limited number of pregnant women and women of childbearing age, without an increase in the frequency of malformation or other direct or indirect harmful effects on the human foetus having been observed. Studies in animals have shown evidence of an increased occurrence of foetal damage, the significance of which is considered uncertain in humans.
There is no information regarding the presence of avatrombopag in human milk, the effects on the breastfed child, or the effects on milk production. Avatrombopag was detected in the milk of lactating rats. A risk to newborns/infants cannot be excluded and breastfeeding is not recommended during treatment with Doptelet and for at least 2 weeks after the last dose.
A lactating woman receiving Doptelet for brief periods, such as prior to an invasive procedure, should interrupt breastfeeding and pump and discard breastmilk during treatment and for two weeks after the last dose of Doptelet in order to minimise exposure to a breastfed child. Advise lactating women receiving chronic Doptelet therapy not to breastfeed during treatment with Doptelet and for at least 2 weeks after the last dose.4.7 Effects on Ability to Drive and Use Machines
Doptelet has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
The following clinically significant adverse events are discussed in detail in other sections of the labelling:
Thrombotic/thromboembolic complications (see Section 4.4 Special Warnings and Precautions for Use).
Clinical trial experience.
Because clinical trials are conducted under widely varying conditions, adverse event rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Patients with chronic liver disease.
The safety of Doptelet was evaluated in two international, identically designed, randomised, double-blind, placebo-controlled trials, ADAPT-1 and ADAPT-2, in which 430 patients with chronic liver disease and thrombocytopenia received either Doptelet (n = 274) or placebo (n = 156) daily for 5 days prior to a scheduled procedure, and had 1 post-dose safety assessment.
Low baseline platelet count cohort (less than 40 x 109/L) who received Doptelet 60 mg once daily for 5 days;
High baseline platelet count cohort (40 to less than 50 x 109/L) who received Doptelet 40 mg once daily for 5 days.
The majority of patients were males (65%) and median subject age was 58 years (ranging from 19-86 years of age). The racial and ethnic distribution was White (60%), Asian (33%), Black (3%), and Other (3%).
The most common adverse events (those occurring in ≥ 3% of patients) in the Doptelet-treated groups (60 mg or 40 mg) across the pooled data from the two trials are summarised in Table 5.
 For the low baseline platelet count cohort, the incidence of serious adverse events was 7% (11/159) in the 60 mg Doptelet treatment group. For the high baseline platelet count cohort, the incidence of serious adverse events was 8% (9/115) in the 40 mg Doptelet treatment group. The most common serious adverse events reported with Doptelet was hyponatremia. Two Doptelet-treated patients (0.7%) developed hyponatremia.
For the low baseline platelet count cohort, the incidence of serious adverse events was 7% (11/159) in the 60 mg Doptelet treatment group. For the high baseline platelet count cohort, the incidence of serious adverse events was 8% (9/115) in the 40 mg Doptelet treatment group. The most common serious adverse events reported with Doptelet was hyponatremia. Two Doptelet-treated patients (0.7%) developed hyponatremia.
Adverse events resulting in discontinuation of Doptelet were anaemia, pyrexia, and myalgia; each was reported in a single (0.4%) patient in the Doptelet (60 mg) treatment group.
Patients with chronic immune thrombocytopenia.
The safety of Doptelet was evaluated in four clinical trials in patients with chronic immune thrombocytopenia: two Phase 3 trials (one randomised, double-blind, placebo-controlled trial, and one randomised, double-blind, active-controlled trial) and two Phase 2 trials (one randomised, double-blind, placebo-controlled, dose-ranging trial, and one open-label extension trial) in 161 patients with chronic immune thrombocytopenia in both the double-blind and open-label extension phases.
The pooled safety data from these four clinical trials includes 128 patients who received 2.5 to 40 mg of Doptelet once daily for a median duration of exposure of 29.1 weeks and had 1 post-dose safety assessment. The majority of patients were female (63%) and median subject age was 50.5 years (ranging from 18-88 years of age). The racial and ethnic distribution was White (84%), Black (6%), Asian (6%) and Other (6%).
The most common adverse events (those occurring in ≥ 10% of patients) in the Doptelet-treated patients across the pooled safety data from the four trials are summarised in Table 6.
 The incidence of serious adverse events was 9% (12/128) in the Doptelet treatment group. Serious adverse events reported in more than 1 individual Doptelet-treated patient included headache, occurring in 1.6% (2/128).
The incidence of serious adverse events was 9% (12/128) in the Doptelet treatment group. Serious adverse events reported in more than 1 individual Doptelet-treated patient included headache, occurring in 1.6% (2/128).
Adverse events resulting in discontinuation of Doptelet that were reported in more than 1 patient included headache, occurring in 1.6% (2/128).
Post-marketing data.
Immune system disorders.
Hypersensitivity reactions including pruritus, rash, choking sensation, erythema, pharyngeal oedema, pruritus generalised, rash macular, swelling face, and swollen tongue.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
In the event of overdose, platelet count may increase excessively and result in thrombotic or thromboembolic complications. Closely monitor the patient and platelet count. Treat thrombotic complications in accordance with standard of care.
No antidote for Doptelet overdose is known.
Haemodialysis is not expected to enhance the elimination of Doptelet because Doptelet is only approximately 6% renally excreted and is highly bound to plasma proteins.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Avatrombopag is an orally bioavailable, small molecule thrombopoietin receptor (TPO-R) agonist that stimulates proliferation and differentiation of megakaryocytes from bone marrow progenitor cells, resulting in an increased production of platelets. Avatrombopag does not compete with TPO for binding to the TPO receptor and has an additive effect with TPO on platelet production.
Pharmacodynamics. Platelet response.
Doptelet administered to adult patients resulted in dose- and exposure-dependent elevations in platelet counts. The onset of the platelet count increase was observed within 3 to 5 days of the start of treatment, with peak effect after 10 to 13 days. Post treatment, platelet counts decreased gradually, returning to near baseline values.
Cardiac electrophysiology.
At exposures similar to that achieved at the 40 mg and 60 mg dose, Doptelet did not prolong the QT interval to any clinically relevant extent. Mean QTc prolongation effects > 20 ms are not anticipated with the highest recommended therapeutic dosing regimen based on analysis of data from the pooled clinical trials in patients with chronic liver disease.
Clinical trials.
Patients with chronic liver disease. The efficacy of Doptelet for the treatment of thrombocytopenia in patients with chronic liver disease who are scheduled to undergo a procedure was established in 2 identically-designed multicentre, randomised, double-blind, placebo-controlled trials (ADAPT-1 and ADAPT-2). In each trial, patients were assigned to the low baseline platelet count cohort (< 40 x 109/L) or the high baseline platelet count cohort (≥ 40 to < 50 x 109/L) based on their platelet count at baseline. Patients were then randomised in a 2:1 ratio to either Doptelet or placebo. Patients were stratified according to hepatocellular cancer (HCC) status and risk of bleeding associated with the elective procedure (low, moderate, or high). Examples of low risk procedures included paracentesis, thoracocentesis, and gastrointestinal endoscopies; examples of moderate risk procedures included liver biopsy, bronchoscopy, and chemoembolisation procedures; and examples of high risk procedures included laparoscopic interventions, transjugular intrahepatic portosystemic shunts, and radiofrequency ablation procedures. Patients undergoing neurosurgical interventions, thoracotomy, laparotomy or organ resection were not eligible for enrolment.
Patients in the low baseline platelet count cohort received 60 mg Doptelet or matching placebo once daily for 5 days, and patients in the high baseline platelet count cohort received 40 mg Doptelet or matching placebo once daily for 5 days. Eligible patients were scheduled to undergo their procedure (low, moderate, or high bleeding risk) 5 to 8 days after their last dose of treatment. Patient populations were similar between the pooled low and high baseline platelet count cohorts and consisted of 66% male and 35% female; median age 58 years and 61% White, 34% Asian, and 3% Black. Patients' MELD scores ranged from < 10 (37.5%), 10 to 14 (46.3%) and from > 14 to < 24 (16.2%), and included patients with CTP Class A (56.4%), Class B (38.1%), and Class C (5.6%).
In ADAPT-1, a total of 231 patients were randomised, 149 patients were treated with Doptelet and 82 patients were treated with placebo. In the low baseline platelet count cohort, the mean baseline platelet count for the Doptelet-treated group was 31.1 x 109/L and for the placebo-treated patients was 30.7 x 109/L. In the high baseline platelet count cohort, the mean baseline platelet count for the Doptelet-treated patients was 44.3 x 109/L and for placebo-treated patients was 44.9 x 109/L.
In ADAPT-2, a total of 204 patients were randomised, 128 patients were treated with Doptelet and 76 patients were treated with placebo. In the low baseline platelet count cohort, the mean baseline platelet count for the Doptelet-treated group was 32.7 x 109/L and for the placebo-treated patients was 32.5 x 109/L. In the high baseline platelet count cohort, the mean baseline platelet count for the Doptelet-treated patients was 44.3 x 109/L and for the placebo-treated patients was 44.5 x 109/L.
Across both baseline platelet count cohorts and the avatrombopag and placebo treatment groups, patients underwent a broad spectrum of types of scheduled procedures that ranged from low to high bleeding risk.
Overall, the majority of patients (60.8% [248/430] subjects) in all treatment groups underwent low bleeding risk procedures, 17.2% (70/430) of patients underwent procedures associated with moderate bleeding risk, and 22.1% (90/430) of subjects underwent procedures associated with high bleeding risk. The proportions of patients undergoing low, moderate, and high-risk procedures were similar between the avatrombopag and placebo treatment groups.
The major efficacy outcome was the proportion of patients who did not require a platelet transfusion or any rescue procedure for bleeding after randomisation and up to 7 days following an elective procedure. Additional secondary efficacy outcomes were the proportion of patients who achieved platelet counts of > 50 x 109/L on the day of procedure, and the change in platelet count from baseline to procedure day.
Responders were defined as patients who did not require a platelet transfusion or any rescue procedure for bleeding after randomisation and up to 7 days following a scheduled procedure. The following were considered rescue therapies to manage the risk of bleeding associated with a procedure: whole blood transfusion, packed red blood cell (RBC) transfusion, platelet transfusion, fresh frozen plasma (FFP) or cryoprecipitate administration, Vitamin K, desmopressin, recombinant activated factor VII, aminocaproic acid, tranexamic acid, or surgical or interventional radiology procedures performed to achieve haemostasis and control blood loss. In both baseline platelet count cohorts, patients in the Doptelet treatment groups had a greater proportion of responders than the corresponding placebo treatment groups that was both clinically meaningful and statistically significant as detailed in Table 7.
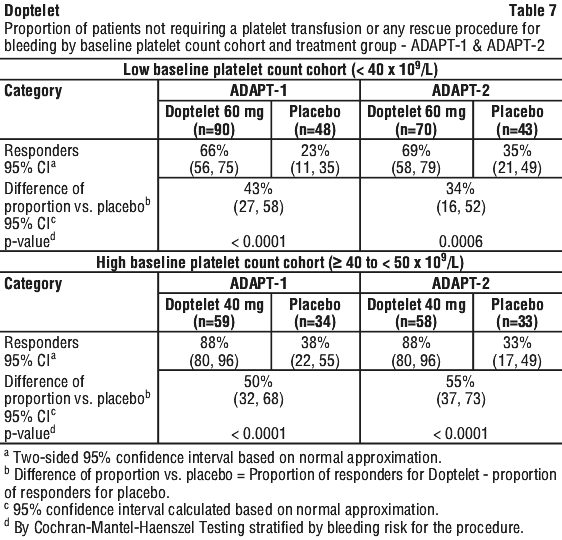 In addition, both trials demonstrated a higher proportion of patients who achieved the target platelet count of ≥ 50 x 109/L on the day of procedure, a secondary efficacy endpoint, in both Doptelet-treated groups versus the placebo-treated groups for both cohorts (low baseline platelet count cohort - ADAPT-1: 69% vs 4%, respectively; p < 0.0001, ADAPT-2: 67% vs 7%, respectively; p < 0.0001; high baseline platelet count cohort - ADAPT-1: 88% vs 21%, respectively; p < 0.0001: ADAPT-2: 93% vs 39%, respectively; p < 0.0001). Further, both trials demonstrated a greater mean change in platelet counts from baseline to the day of the procedure, a secondary efficacy endpoint, in both Doptelet-treated groups versus the placebo-treated groups for both cohorts (low baseline platelet count cohort - ADAPT-1: 32 x 109/L vs 0.8 x 109/L, respectively; p < 0.0001; ADAPT-2: 31.3 x 109/L vs 3.0 x 109/L, respectively; p < 0.0001; high baseline platelet count cohort - ADAPT-1: 37.1 x 109/L vs 1.0 x 109/L, respectively; p < 0.0001; ADAPT-2: 44.9 x 109/L vs 5.9 x 109/L, respectively; p < 0.0001).
In addition, both trials demonstrated a higher proportion of patients who achieved the target platelet count of ≥ 50 x 109/L on the day of procedure, a secondary efficacy endpoint, in both Doptelet-treated groups versus the placebo-treated groups for both cohorts (low baseline platelet count cohort - ADAPT-1: 69% vs 4%, respectively; p < 0.0001, ADAPT-2: 67% vs 7%, respectively; p < 0.0001; high baseline platelet count cohort - ADAPT-1: 88% vs 21%, respectively; p < 0.0001: ADAPT-2: 93% vs 39%, respectively; p < 0.0001). Further, both trials demonstrated a greater mean change in platelet counts from baseline to the day of the procedure, a secondary efficacy endpoint, in both Doptelet-treated groups versus the placebo-treated groups for both cohorts (low baseline platelet count cohort - ADAPT-1: 32 x 109/L vs 0.8 x 109/L, respectively; p < 0.0001; ADAPT-2: 31.3 x 109/L vs 3.0 x 109/L, respectively; p < 0.0001; high baseline platelet count cohort - ADAPT-1: 37.1 x 109/L vs 1.0 x 109/L, respectively; p < 0.0001; ADAPT-2: 44.9 x 109/L vs 5.9 x 109/L, respectively; p < 0.0001).
A measured increase in platelet counts was observed in both Doptelet treatment groups over time beginning on day 4 post-dose, that peaked on day 10-13, decreased 7 days post-procedure, and then returned to near baseline values by day 35.
Patients with chronic immune thrombocytopenia. Randomised phase 3 clinical trial.
The efficacy of Doptelet in adult patients with chronic immune thrombocytopenia was evaluated in a phase 3, multicentre, randomised, double-blind, placebo-controlled trial. Patients had previously received one or more prior chronic immune thrombocytopenia therapies and had an average of screening and baseline platelet counts < 30 x 109/L. Patients were centrally stratified by splenectomy status, baseline platelet count (≤ 15 x 109/L or > 15 x 109/L to < 30 x 109/L), and use of concomitant chronic immune thrombocytopenia medication, and then randomised (2:1) to receive either Doptelet or placebo for 6 months. Patients received a starting dose of 20 mg once daily, with doses subsequently titrated based on platelet response.
Forty-nine patients were randomised, 32 to Doptelet and 17 to placebo, with similar mean [SD] baseline platelet counts in the 2 treatment groups (14.1 [8.6] x 109/L and 12.7 [7.8] x 109/L, respectively). The median age was 44 years, 63% were female, and 94% were Caucasian, 4% Asian and 2% Black. The median duration of exposure was 26 weeks for Doptelet-treated patients and 6 weeks for placebo-treated patients. The major efficacy outcome in this trial was the cumulative number of weeks in which the platelet count was ≥ 50 x 109/L during the 6-month treatment period in the absence of rescue therapy. Doptelet-treated patients had a longer duration of platelet counts ≥ 50 x 109/L in the absence of rescue therapy than those who received placebo (median 12.4 [0, 25] vs 0 [0, 2] weeks, respectively, p < 0.0001) (see Table 8).
 In addition, a larger proportion of patients in the Doptelet treatment group had platelet counts ≥ 50 x 109/L at day 8 compared to placebo (21/32; 66% vs 0/17; 0.0%, respectively; p < 0.0001).
In addition, a larger proportion of patients in the Doptelet treatment group had platelet counts ≥ 50 x 109/L at day 8 compared to placebo (21/32; 66% vs 0/17; 0.0%, respectively; p < 0.0001).
5.2 Pharmacokinetic Properties
Avatrombopag demonstrated dose-proportional pharmacokinetics after single doses from 10 mg (0.5 times the lowest approved dosage) to 80 mg (1.3 times the highest recommended dosage). Healthy subjects administered 40 mg of avatrombopag had a geometric mean (%CV) maximal concentration (Cmax) of 166 (84%) nanogram/mL and area under the time-concentration curve extrapolated to infinity (AUC0-inf) of 4198 (83%) nanogram.hr/mL. The pharmacokinetics of avatrombopag were similar in both healthy subjects and the chronic liver disease population.
Absorption.
The median time to maximal concentration (Tmax) occurred at 5 to 6 hours post-dose in the fasted state.
Effect of food.
Avatrombopag AUC0-inf and Cmax were not affected when Doptelet was co-administered with a low-fat meal (500 calories, 3 g fat, 15 g protein, and 108 g carbohydrates) or a high fat meal (918 calories, 59 g fat, 39 g protein, and 59 g carbohydrates). The variability of avatrombopag exposure was reduced by 40% to 60% with food. The Tmax of avatrombopag was delayed by 0 to 2 hours when Doptelet was administered with a low-fat or high-fat meal (median Tmax range 5 to 8 hours) compared to the fasted state.
Distribution.
Avatrombopag has an estimated mean volume of distribution (%CV) of 180 L (25%). Avatrombopag is greater than 96% bound to human plasma proteins.
Metabolism.
Avatrombopag is primarily metabolised by cytochrome P450 CYP2C9 and CYP3A4.
Excretion.
The mean plasma elimination half-life (%CV) of avatrombopag is approximately 19 hours (19%). The mean (%CV) of the clearance of avatrombopag is estimated to be 6.9 L/hr (29%).
Faecal excretion accounted for 88% of the administered dose, with 34% of the dose excreted as unchanged avatrombopag. Only 6% of the administered dose was found in urine.
Specific populations.
Age (18-86 years), body weight (39-175 kg), sex, race [Whites, African-Americans, and East Asians (i.e. Japanese, Chinese and Koreans)], and any hepatic impairment (Child-Turcotte-Pugh (CTP) grade A, B, and C, or Model for End-Stage Liver Disease (MELD) score 4-23) and mild to moderate renal impairment (CLcr ≥ 30 mL/min) did not have clinically meaningful effects on the pharmacokinetics of avatrombopag.
The effect of age (< 18 years) and severe renal impairment (CLcr < 30 mL/min, Cockcroft-Gault) including patients requiring haemodialysis on avatrombopag pharmacokinetics is unknown.
Drug interactions.
Clinical studies.
Table 9 summarises the effect of other drugs on the pharmacokinetics of avatrombopag.

5.3 Preclinical Safety Data
Genotoxicity.
Avatrombopag was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay or clastogenic in an in vitro human lymphocyte chromosomal aberrations assay or in an in vivo rat bone marrow micronucleus assay.
Carcinogenicity.
In two-year carcinogenicity studies, avatrombopag was administered orally at doses of 20, 60, 160 mg/kg/day in mice and doses of 20, 50, 160 mg/kg/day in rats. Avatrombopag induced a statistically significant increase in neuroendocrine cell (enterochromaffin-like cell, ECL cell) gastric tumours (carcinoids) in the stomach at 160 mg/kg in female rats at 117 and 81 times the clinical exposure based on the AUC in patients at the maximum recommended dose of 60 mg (CLD) and 40 mg (ITP), respectively, once daily. The gastric carcinoids were considered likely due to prolonged hypergastrinemia observed in toxicity studies. Hypergastrinemia-related gastric carcinoids in rodents are generally considered to be of low risk or relevance to humans.
However, avatrombopag activates TPO receptors on the surface of haematopoietic cells. TPO has been shown to stimulate the proliferation of megakaryocytic leukaemia cells in vitro. There is therefore a theoretical possibility that avatrombopag may increase the risk for haematologic malignancies.6 Pharmaceutical Particulars
6.1 List of Excipients
Tablet core.
Lactose monohydrate, colloidal anhydrous silica, crospovidone, magnesium stearate and microcrystalline cellulose.
Film coating.
Opadry II Complete Film Coating System 85F42244 Yellow.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C.
6.5 Nature and Contents of Container
Doptelet is supplied in PA/Al/PVC/Al blister packs of 10, 15 or 30 tablets.
Not all pack sizes may be marketed.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Avatrombopag maleate is a white to off-white non-hygroscopic powder, practically insoluble in water at all pH values. Partition coefficient of avatrombopag maleate is > 4.0 (log D at pH 3-9), and pKa values of avatrombopag maleate are 2.8, 3.6 and 8.4.
Chemical structure.

CAS number.
677007-74-8.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes

 Doptelet has been investigated only as a single 5-day once daily dosing regimen in clinical trials in patients with chronic liver disease (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Doptelet should not be administered to patients with chronic liver disease in an attempt to normalise platelet counts.
Doptelet has been investigated only as a single 5-day once daily dosing regimen in clinical trials in patients with chronic liver disease (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Doptelet should not be administered to patients with chronic liver disease in an attempt to normalise platelet counts.
 In the case of a missed dose, patients should take the missed dose of Doptelet as soon as they remember. Patients should not take two doses at one time to make up for a missed dose and should take the next dose per the current regimen.
In the case of a missed dose, patients should take the missed dose of Doptelet as soon as they remember. Patients should not take two doses at one time to make up for a missed dose and should take the next dose per the current regimen.