What is in this leaflet
This leaflet answers some common questions about Dupixent.
It does not contain all the available information. It does not take the place of talking to your Doctor or Pharmacist.
All medicines have risks and benefits. Your Doctor has weighed the risks of you using Dupixent against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your Doctor or Pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What DUPIXENT is used for
Dupixent contains the active substance dupilumab.
Dupixent is used to treat the following conditions:
- moderate to severe atopic dermatitis (also known as atopic eczema), in patients aged 12 years and older.
In atopic dermatitis, Dupixent may be used with or without prescribed atopic dermatitis medicines that you apply to the skin.
- maintenance treatment of moderate-to-severe asthma in patients 12 year and older whose asthma is not controlled with their current asthma medicines.
In asthma, Dupixent should be used in addition to your existing asthma medicines. Dupixent may also help reduce the amount of oral corticosteroids you need while preventing severe asthma attacks and improving your breathing.
Your doctor will assess if Dupixent is appropriate for your condition.
How it works
Dupixent is an injectable medicine that inhibits IL-4 and IL-13 proteins by blocking a shared receptor. IL-4 and IL-13 play a major role in the symptoms of atopic dermatitis and asthma.
Dupixent belongs to a class of medicines called monoclonal antibodies. Monoclonal antibodies are proteins that specifically recognise and bind to other unique proteins in the body.
Ask your Doctor if you have any questions about why Dupixent has been prescribed for you.
Before you use DUPIXENT
When you must not use Dupixent
Do not use Dupixent if you have an allergy to:
- any medicine containing dupilumab (the active ingredient) or any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- rash, itching or hives on the skin
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
Tell your doctor if you are experiencing these symptoms
Dupixent should not be used after the expiry date (exp) printed on the pack. If you use this medicine after the expiry date has passed, it may not work as well, or have an unexpected effect.
Dupixent should not be used if the packaging is torn or shows signs of tampering.
Do not use Dupixent if the product appears cloudy, discoloured or contains particles, or if the syringe and/or needle cap appear damaged.
Atopic Dermatitis:
Do not use Dupixent in patients under 12 years old. The safety and benefits of Dupixent are not yet known in this patient group.
Asthma:
Do not use Dupixent in patients under 12 years old. The safety and benefits of Dupixent are not yet known in this patient group.
Dupixent is not a rescue medicine and should not be used to treat a sudden asthma attack.
If you are not sure whether you should start using this medicine, talk to your Doctor.
Before you start to use DUPIXENT
Tell your doctor or pharmacist if you have allergies to any other medicines, or substances such as foods, preservatives or dyes.
Tell your Doctor if you have any of the following, or if any of the following apply to you:
- have a parasitic (helminth) infection
- are pregnant or plan to become pregnant. It is not known if Dupixent will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known whether Dupixent passes into breast milk.
- have recently received or are scheduled to receive a vaccine
- any new or worsening eye problems, including eye pain or changes in vision.
- are taking oral, or using topical, or inhaled corticosteroid medicines
- have any other medical conditions
If you have not told your Doctor about any of the above, tell them before you start using Dupixent.
Taking other medicines
Tell your Doctor or pharmacist if you are taking any other medicines, have recently taken, or might take any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Do not stop taking your corticosteroid medicines unless instructed by your doctor. This may cause other symptoms that were controlled by the corticosteroid medicine to come back.
You should not receive certain type of vaccines while taking Dupixent. Tell your Doctor if you have recently received a vaccine or planned to receive a vaccine.
How to use DUPIXENT
Dupixent comes as a single-dose (1 time use) pre-filled syringe with or without needle shield. Your healthcare provider will prescribe the type that is best for you. Follow all directions given to you by your doctor and pharmacist carefully.
Carefully read the "Dupixent Instructions for Use" provided in the carton. If you do not understand the instructions on the label or in this leaflet, ask your doctor or pharmacist for help.
How to use it
Always check the syringe label before each injection to make sure you are using the right medicine.
Dupixent is a clear and colourless to pale yellow solution that should not be shaken before use. Do not use Dupixent if it is not clear to pale yellow or if it contains particles.
To avoid discomfort, Dupixent should be removed from the refrigerator before your injection so that it reaches room temperature.
- Remove Dupixent 300mg syringes from the refrigerator at least 45 minutes before injection.
- Remove Dupixent 200mg syringes from the refrigerator at least 30 minutes before injection.
The syringe should not be exposed to heat or direct sunlight.
The injection can be self-administered or given by another person, after proper training in injection technique.
Dupixent syringes are pre-filled and ready to use. Once the contents have been injected, the syringe cannot be re-used.
Never use a syringe if it is damaged, or you are not sure that it is working properly. Use a new syringe.
After removal from the refrigerator, Dupixent must be used within 14 days or discarded.
Dupixent is intended for injection under the skin.
If you do not understand the instructions, ask your Doctor or Pharmacist.
How much to use
Use the dose that your doctor prescribes for you.
Dupixent is given as an injection under the skin (subcutaneously) once every two weeks.
Where to inject
Dupixent is injected under the skin (subcutaneous injection). You can inject into the thigh or abdomen, except for the 2 inches (5 cm) around the navel, using a single-dose pre-filled syringe. If somebody else administers the injection, the upper arm can also be used. Use a different injection site each injection so that the same site is not used.
Do not inject in an area where the skin that is tender, damaged, sunburnt or has bruises or scars.
Do not inject Dupixent with other injectable medicines, at the same injection site.
How long to use Dupixent
Continue using Dupixent for as long as your doctor recommends.
Make sure you keep enough Dupixent to last when you go on holidays.
If you forget to use it
If you miss a dose of DUPIXENT, give the injection within 7 days from the missed dose, then continue with the original schedule.
If the missed dose is not given within 7 days, wait until the next scheduled dose to give your DUPIXENT injection.
In case you are not sure when to inject Dupixent, call your doctor or pharmacist.
It is important to use Dupixent as prescribed by your doctor.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (13 11 26) or go to accident and emergency at your nearest hospital, if you think that you have taken too much Dupixent. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Always take the outer carton of the medicine with you.
While you are using DUPIXENT
If you experience any symptoms of an allergic reaction, stop using Dupixent and talk to your doctor immediately.
Things you must do
Always follow your doctor’s instructions carefully.
Keep Dupixent in a refrigerator (2°C - 8°C). Do not freeze. Do not expose to extreme heat.
Dupixent should be removed from the refrigerator before your injection to avoid discomfort.
- Dupixent 300 mg syringes should be removed from the refrigerator at least 45 minutes before your injection.
- Dupixent 200 mg syringes should be removed from the refrigerator at least 30 minutes before your injection.
Dupixent should not be exposed to heat or direct sunlight.
After removal from the refrigerator, Dupixent must be used within 14 days or discarded.
It is important to keep using Dupixent even if you feel well.
Continuous use of Dupixent helps to control your condition.
If you become pregnant while you are using Dupixent, tell your doctor.
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
If you have asthma and are taking asthma medicines, do not change or stop your asthma medicine without talking to your doctor.
If you are about to be started on any new medicine, tell your doctor and pharmacist that you are taking Dupixent.
Things you must not do
Do not use Dupixent if you think it has been frozen or exposed to excessive heat (temperatures above 25°C).
Do not give Dupixent to anyone else, even if they have the same condition as you.
Do not stop taking your corticosteroid medicines unless instructed by your doctor. This may cause other symptoms that were controlled by the corticosteroid medicine to come back
Do not stop taking your medicine without checking with your doctor.
Things to be aware of
Dupixent is unlikely to influence your ability to drive and use machines.
Side effects
Tell your Doctor as soon as possible if you do not feel well while you are using Dupixent.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your Doctor or Pharmacist to answer any questions you may have.
Tell your doctor if you notice any of them and they worry you or it does not go away:
- Injection site reactions
- headache
- Eye dryness, eye infection, eye and eyelid inflammation (including redness/swelling/ itching) (if you also have atopic dermatitis)
- eye pain
- blurred vision
- sensitivity to light
- oral herpes (cold sores)
- a specific form of allergic reaction resulting in inflammation of your blood vessels. Rarely, this can happen in people with asthma who receive Dupixent.
In rare cases, patients taking an asthma medicine may develop an inflammation of blood vessels or lungs. It is not known whether this is caused by Dupixent. This usually, but not always, happens in people who also take a steroid medicine which is being stopped or for which the dose is being lowered.
Tell your doctor immediately if you develop a combination of the following symptoms:
- a flu-like illness
- pins and needles or numbness of arms or legs
- worsening of lung symptoms including breathing problems,
- and/or rash.
Dupixent can cause serious side effects, including: generalized allergic (hypersensitivity) reactions including anaphylaxis can happen after you get your Dupixent injection.
If you have any signs of allergic (hypersensitivity) reaction stop using Dupixent, tell your Doctor immediately or go to Accident and Emergency at your nearest hospital.
Symptoms of allergic reactions include:
- shortness of breath
- persistent fever
- chest pain
- general ill feeling
- swollen lymph nodes
- swelling of the face, mouth, and tongue
- hives
- itching
- fainting, dizziness, feeling lightheaded (low blood pressure)
- joint pain
- skin rash
- a feeling of pins and needles or numbness of your arms or legs
These are not all of the possible side effects of Dupixent. Call your Doctor for medical advice about side effects.
Other side effects not listed above may also occur in some patients. Tell your doctor if you notice anything else that is making you feel unwell.
After using DUPIXENT
Storage
All medicines should be kept where children cannot reach them.
Before use, keep Dupixent syringes in a refrigerator where the temperature is between 2-8°C. If necessary, pre-filled syringes can be kept at room temperature up to 25°C for a maximum of 14 days.
Keep the syringes in the original carton to protect from light.
Do not allow it to freeze. Discard if frozen.
Do not expose to heat. Do not shake.
If you need to travel, make sure the medicine is kept at the right temperature. This is important whether travelling by car, bus, train, plane or any other form of transport.
Disposal
After injecting Dupixent, immediately throw away the used pre-filled syringe in a sharps container as instructed by your doctor or pharmacist.
If your Doctor tells you to stop using Dupixent or the expiry date has passed, ask your Pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Dupixent is a clear and colourless to pale yellow, preservative-free liquid solution available in a pre-filled syringe with or without needle shield.
The needle shield reduces the risk of accidental needle stick injuries.
Dupixent is available in two different strengths:
- 300 mg in 2 mL solution
- 200 mg in 1.14 mL solution
Ingredients
Active Ingredient:
- dupilumab
Inactive Ingredients:
- acetic acid
- arginine hydrochloride
- histidine
- polysorbate 80
- sodium acetate
- sucrose
- water for injections
Supplier
Dupixent is supplied in Australia by:
sanofi-aventis australia pty ltd
12-24 Talavera Road
Macquarie Park NSW 2113
Freecall No: 1800 818 806
sanofi-aventis new zealand limited
Level 8, James and Wells Tower
56 Cawley Street, Ellerslie,
Auckland
New Zealand
Freecall No:0800 283 684
Dupixent 300 mg (150 mg/mL) pre-filled syringe with needle shield
AUST R 283127
Dupixent 300 mg (150mg/mL) pre-filled syringe
AUST R 282981
Dupixent 200 mg (175 mg/mL) pre-filled syringe with needle shield
AUST R 302463
This leaflet was prepared in October 2020
dupi-ccdsv13-cmiv8-06oct20
Published by MIMS November 2020

 The Dupixent pre-filled pen is for use in adult and paediatric patients aged 2 years and older.
The Dupixent pre-filled pen is for use in adult and paediatric patients aged 2 years and older. For children (6-11 years old) with asthma and co-morbid severe atopic dermatitis, the recommended dose should be followed in Table 1.
For children (6-11 years old) with asthma and co-morbid severe atopic dermatitis, the recommended dose should be followed in Table 1. Table 5 summarises the adverse reactions that occurred in ≥ 1% of patients treated with Dupixent during the first 16-weeks of treatment in placebo-controlled trials.
Table 5 summarises the adverse reactions that occurred in ≥ 1% of patients treated with Dupixent during the first 16-weeks of treatment in placebo-controlled trials. The safety profile of Dupixent + TCS through week 52 is consistent with the safety profile observed at week 16.
The safety profile of Dupixent + TCS through week 52 is consistent with the safety profile observed at week 16. In PRIME and PRIME2 PN studies, the proportion of patients who discontinued treatment due to adverse events was 0% of the dupilumab 300 mg Q2W group and 2.5% of the placebo group.
In PRIME and PRIME2 PN studies, the proportion of patients who discontinued treatment due to adverse events was 0% of the dupilumab 300 mg Q2W group and 2.5% of the placebo group. See Table 8.
See Table 8. The long-term safety of Dupixent was assessed in an open-label extension study in 2282 patients 12 years and older with moderate-to-severe asthma (TRAVERSE). In this study, patients were followed for up to 96 weeks, resulting in 3169 patient-years cumulative exposure to Dupixent. The safety profile of Dupixent in TRAVERSE was consistent with the safety profile observed in pivotal asthma studies for up to 52 weeks of treatment. No additional adverse reactions were identified.
The long-term safety of Dupixent was assessed in an open-label extension study in 2282 patients 12 years and older with moderate-to-severe asthma (TRAVERSE). In this study, patients were followed for up to 96 weeks, resulting in 3169 patient-years cumulative exposure to Dupixent. The safety profile of Dupixent in TRAVERSE was consistent with the safety profile observed in pivotal asthma studies for up to 52 weeks of treatment. No additional adverse reactions were identified. See Table 10.
See Table 10. The safety profile of Dupixent through Week 52 was generally consistent with the safety profile observed at Week 24.
The safety profile of Dupixent through Week 52 was generally consistent with the safety profile observed at Week 24.


 Treatment effects in subgroups (weight, age, gender, race, and background treatment, including immunosuppressants) in Study 1334 and Study 1416 were in general consistent with the results in the overall study population.
Treatment effects in subgroups (weight, age, gender, race, and background treatment, including immunosuppressants) in Study 1334 and Study 1416 were in general consistent with the results in the overall study population.

 Treatment effects in evaluable subgroups (weight, age, gender, race, and background treatment, including immunosuppressants) in Study 1224 were in general consistent with the results in the overall study population.
Treatment effects in evaluable subgroups (weight, age, gender, race, and background treatment, including immunosuppressants) in Study 1224 were in general consistent with the results in the overall study population. In SOLO CONTINUE, a trend for increased treatment-emergent ADA positivity with increased dosing intervals was observed. Treatment-emergent ADA: QW: 1.2%; Q2W: 4.3%; Q4W: 6.0%; Q8W: 11.7%. ADA responses lasting more than 12 weeks: QW: 0.0%; Q2W: 1.4%; Q4W: 0.0%; Q8W: 2.6%.
In SOLO CONTINUE, a trend for increased treatment-emergent ADA positivity with increased dosing intervals was observed. Treatment-emergent ADA: QW: 1.2%; Q2W: 4.3%; Q4W: 6.0%; Q8W: 11.7%. ADA responses lasting more than 12 weeks: QW: 0.0%; Q2W: 1.4%; Q4W: 0.0%; Q8W: 2.6%.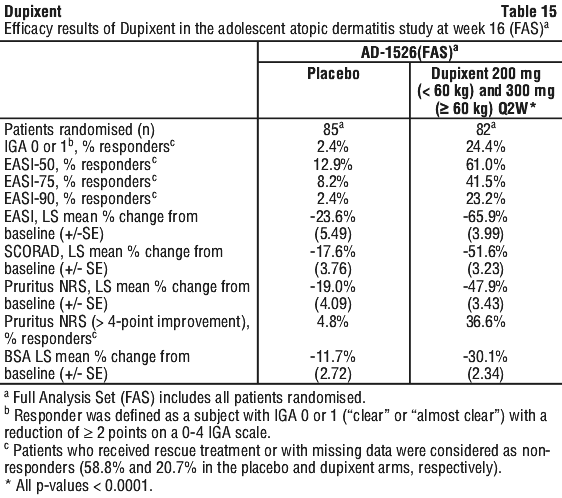 A larger percentage of patients randomised to placebo needed rescue treatment (topical corticosteroids, systemic corticosteroids, or systemic non-steroidal immunosuppressants) as compared to the Dupixent group (58.8% and 20.7%, respectively).
A larger percentage of patients randomised to placebo needed rescue treatment (topical corticosteroids, systemic corticosteroids, or systemic non-steroidal immunosuppressants) as compared to the Dupixent group (58.8% and 20.7%, respectively).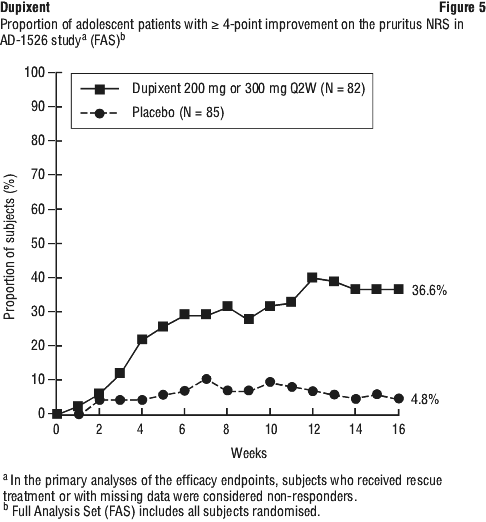 The long-term efficacy of Dupixent in adolescent patients with moderate-to-severe AD who had participated in previous clinical trials of Dupixent was assessed in open-label extension study (AD-1434). Efficacy data from this study suggests that clinical benefit provided at week 16 was sustained through week 52.
The long-term efficacy of Dupixent in adolescent patients with moderate-to-severe AD who had participated in previous clinical trials of Dupixent was assessed in open-label extension study (AD-1434). Efficacy data from this study suggests that clinical benefit provided at week 16 was sustained through week 52. A greater proportion of patients randomised to Dupixent + TCS achieved an improvement in the peak pruritus NRS compared to placebo + TCS (defined as ≥ 4-point improvement at week 4). See Figure 6.
A greater proportion of patients randomised to Dupixent + TCS achieved an improvement in the peak pruritus NRS compared to placebo + TCS (defined as ≥ 4-point improvement at week 4). See Figure 6. The Dupixent groups significantly improved patient-reported symptoms, the impact of AD on sleep and health-related quality of life as measured by POEM, SCORAD, and CDLQI scores at 16 weeks compared to placebo.
The Dupixent groups significantly improved patient-reported symptoms, the impact of AD on sleep and health-related quality of life as measured by POEM, SCORAD, and CDLQI scores at 16 weeks compared to placebo. A significantly greater proportion of patients randomised to dupilumab + TCS achieved a rapid improvement in the Worst Scratch/Itch NRS compared to placebo + TCS (defined as ≥ 4-point improvement as early as week 3, nominal p < 0.005) and the proportion of patients responding on the Worst Scratch/Itch NRS continued to increase through the treatment period (see Figure 7).
A significantly greater proportion of patients randomised to dupilumab + TCS achieved a rapid improvement in the Worst Scratch/Itch NRS compared to placebo + TCS (defined as ≥ 4-point improvement as early as week 3, nominal p < 0.005) and the proportion of patients responding on the Worst Scratch/Itch NRS continued to increase through the treatment period (see Figure 7). In this study, Dupixent significantly improved health-related quality of life as measured by the CDLQI (in 85 patients 4 to 5 years old) and IDQOL (in 77 patients 6 months to 3 years old). In the ITT population, greater LS mean changes in CDLQI and IDQOL scores from baseline to week 16 were observed in the dupilumab + TCS (-10.0 and -10.9) group compared to the placebo + TCS group (-2.5 and -2.0), respectively (p < 0.0001). Similar improvements in both CDLQI and IDQOL were observed in the severe AD population.
In this study, Dupixent significantly improved health-related quality of life as measured by the CDLQI (in 85 patients 4 to 5 years old) and IDQOL (in 77 patients 6 months to 3 years old). In the ITT population, greater LS mean changes in CDLQI and IDQOL scores from baseline to week 16 were observed in the dupilumab + TCS (-10.0 and -10.9) group compared to the placebo + TCS group (-2.5 and -2.0), respectively (p < 0.0001). Similar improvements in both CDLQI and IDQOL were observed in the severe AD population. The onset of action in change from baseline in WI-NRS, defined as the first timepoint at which difference from placebo was and remained significant (nominal p < 0.05) in the weekly average of daily WI-NRS, was observed as early as Week 3 in PRIME (Figure 8) and Week 4 in PRIME 2 (Figure 9).
The onset of action in change from baseline in WI-NRS, defined as the first timepoint at which difference from placebo was and remained significant (nominal p < 0.05) in the weekly average of daily WI-NRS, was observed as early as Week 3 in PRIME (Figure 8) and Week 4 in PRIME 2 (Figure 9).
 A greater proportion of patients experienced WI-NRS improvements of ≥ 4 points from baseline by Weeks 4 and 11 in the dupilumab group as compared to the placebo group in PRIME (Figure 10, nominal p < 0.007) and PRIME2 (Figure 11, nominal p < 0.013) respectively and this difference remained significant throughout the treatment period.
A greater proportion of patients experienced WI-NRS improvements of ≥ 4 points from baseline by Weeks 4 and 11 in the dupilumab group as compared to the placebo group in PRIME (Figure 10, nominal p < 0.007) and PRIME2 (Figure 11, nominal p < 0.013) respectively and this difference remained significant throughout the treatment period.
 Treatment effects on both pruritus and lesions in subgroups (weight, age, gender, race, medical history of atopy, prior use of immunosuppressants and neuromodulators, and concomitant treatment with TCS) were consistent with the results at Week 24 in the overall study population.
Treatment effects on both pruritus and lesions in subgroups (weight, age, gender, race, medical history of atopy, prior use of immunosuppressants and neuromodulators, and concomitant treatment with TCS) were consistent with the results at Week 24 in the overall study population.
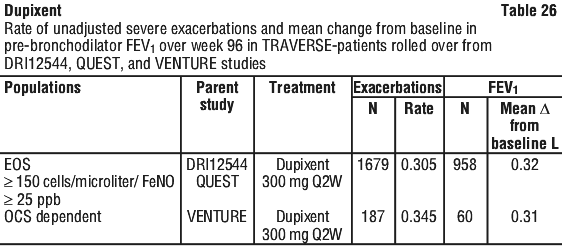
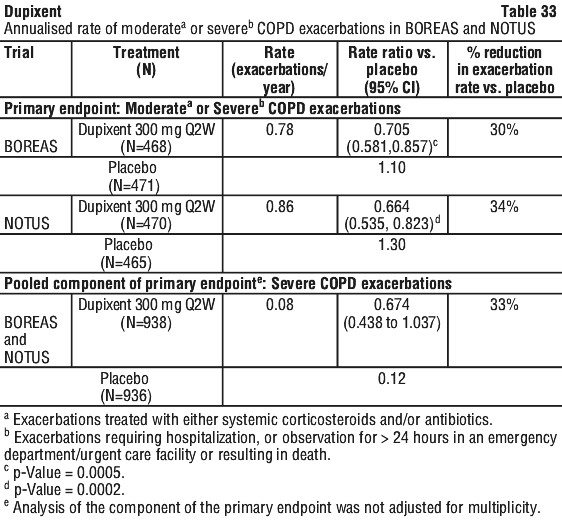
 Prespecified subgroup analyses of DRI12544, QUEST, and VENTURE studies demonstrated that there were greater reductions in severe exacerbations in patients with higher baseline levels of markers for Type 2 inflammation such as eosinophil level and FeNO.
Prespecified subgroup analyses of DRI12544, QUEST, and VENTURE studies demonstrated that there were greater reductions in severe exacerbations in patients with higher baseline levels of markers for Type 2 inflammation such as eosinophil level and FeNO.
 The cumulative mean number of severe exacerbation events in DRI12544, QUEST, and VENTURE studies (Overall Population and Baseline Eosinophils ≥ 300 cells/microL) during the 24- or 52-week treatment period is shown in Figure 13.
The cumulative mean number of severe exacerbation events in DRI12544, QUEST, and VENTURE studies (Overall Population and Baseline Eosinophils ≥ 300 cells/microL) during the 24- or 52-week treatment period is shown in Figure 13. Over the course of the studies, patients in both Dupixent dose groups had lower cumulative number of events compared with patients in their respective placebo groups.
Over the course of the studies, patients in both Dupixent dose groups had lower cumulative number of events compared with patients in their respective placebo groups.

 Significant improvements in FEV1 were observed as early as Week 2 (DRI12544, QUEST, and VENTURE) following the first dose of Dupixent for both the 200 mg and 300 mg dose strengths and were maintained through Week 24 (DRI12544 and VENTURE) and Week 52 (QUEST) (see Figure 15).
Significant improvements in FEV1 were observed as early as Week 2 (DRI12544, QUEST, and VENTURE) following the first dose of Dupixent for both the 200 mg and 300 mg dose strengths and were maintained through Week 24 (DRI12544 and VENTURE) and Week 52 (QUEST) (see Figure 15).


 Exacerbations were defined as deterioration of asthma requiring the use of systemic corticosteroids for at least 3 days or hospitalization or emergency room visit due to asthma that required systemic corticosteroids. Dupixent significantly reduced the annualized rate of severe asthma exacerbation events during the 52-week treatment period compared to placebo in the population with type 2 inflammation and in population defined by baseline blood eosinophils ≥ 300 cells/microL or by baseline FeNO ≥ 20 ppb. Clinically significant improvements in percent predicted pre-bronchodilator FEV1 were observed at Week 12. Improvements were also observed for ACQ-7-IA and PAQLQ(S)-IA at Week 24 and were sustained at Week 52. Greater responder rates were observed for ACQ-7-IA and PAQLQ(S)-IA compared to placebo at Week 24. The efficacy results for VOYAGE are presented in Table 28.
Exacerbations were defined as deterioration of asthma requiring the use of systemic corticosteroids for at least 3 days or hospitalization or emergency room visit due to asthma that required systemic corticosteroids. Dupixent significantly reduced the annualized rate of severe asthma exacerbation events during the 52-week treatment period compared to placebo in the population with type 2 inflammation and in population defined by baseline blood eosinophils ≥ 300 cells/microL or by baseline FeNO ≥ 20 ppb. Clinically significant improvements in percent predicted pre-bronchodilator FEV1 were observed at Week 12. Improvements were also observed for ACQ-7-IA and PAQLQ(S)-IA at Week 24 and were sustained at Week 52. Greater responder rates were observed for ACQ-7-IA and PAQLQ(S)-IA compared to placebo at Week 24. The efficacy results for VOYAGE are presented in Table 28. Response rates by baseline blood eosinophils and FeNO for VOYAGE are shown in Figure 16.
Response rates by baseline blood eosinophils and FeNO for VOYAGE are shown in Figure 16. Improvements in percent predicted FEV1 by baseline blood eosinophils and FeNO for VOYAGE are shown in Figure 17.
Improvements in percent predicted FEV1 by baseline blood eosinophils and FeNO for VOYAGE are shown in Figure 17. Significant improvements in percent predicted FEV1 were observed as early as Week 2 and were maintained through Week 52 in VOYAGE study.
Significant improvements in percent predicted FEV1 were observed as early as Week 2 and were maintained through Week 52 in VOYAGE study. In VOYAGE, in the population with type 2 inflammation, the mean annualized total number of systemic corticosteroid courses due to asthma was reduced by 59.3% versus placebo (0.350 [95% CI: 0.256, 0.477] versus 0.860 [95% CI: 0.616, 1.200]). In the population defined by baseline blood eosinophils > 300 cells/microL, the mean annualized total number of systemic corticosteroid courses due to asthma was reduced by 66.0% versus placebo (0.274 [95% CI: 0.188, 0.399] versus 0.806 [95% CI: 0.563, 1.154]).
In VOYAGE, in the population with type 2 inflammation, the mean annualized total number of systemic corticosteroid courses due to asthma was reduced by 59.3% versus placebo (0.350 [95% CI: 0.256, 0.477] versus 0.860 [95% CI: 0.616, 1.200]). In the population defined by baseline blood eosinophils > 300 cells/microL, the mean annualized total number of systemic corticosteroid courses due to asthma was reduced by 66.0% versus placebo (0.274 [95% CI: 0.188, 0.399] versus 0.806 [95% CI: 0.563, 1.154]).
 The results of SINUS-52 study at week 52 are presented in Table 31.
The results of SINUS-52 study at week 52 are presented in Table 31.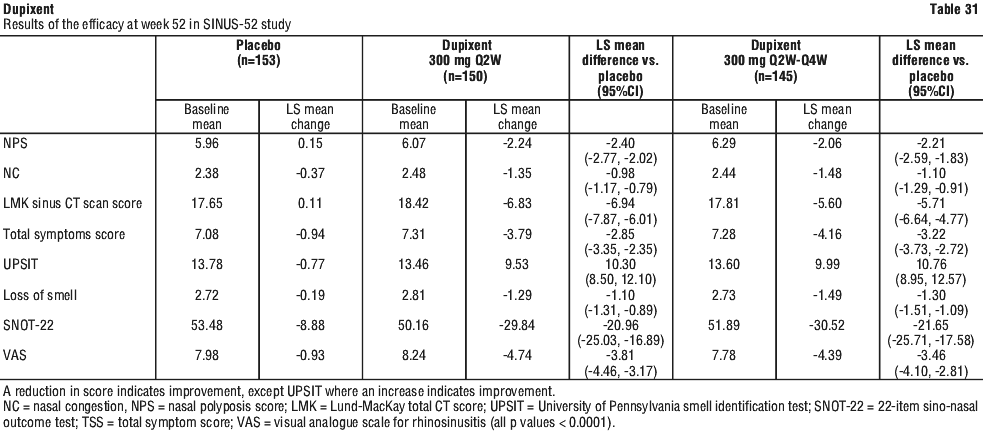 Statistically significant and clinically meaningful efficacy was observed in SINUS-24 with regard to improvement in bilateral endoscopic NPS score at week 24. In the post-treatment period when patients were off dupilumab, the treatment effect diminished over time (see Figure 19).
Statistically significant and clinically meaningful efficacy was observed in SINUS-24 with regard to improvement in bilateral endoscopic NPS score at week 24. In the post-treatment period when patients were off dupilumab, the treatment effect diminished over time (see Figure 19). Statistically significant and clinically meaningful results were also seen in SINUS-52 at both week 24 and week 52 with a progressive improvement over time (see Figure 20).
Statistically significant and clinically meaningful results were also seen in SINUS-52 at both week 24 and week 52 with a progressive improvement over time (see Figure 20).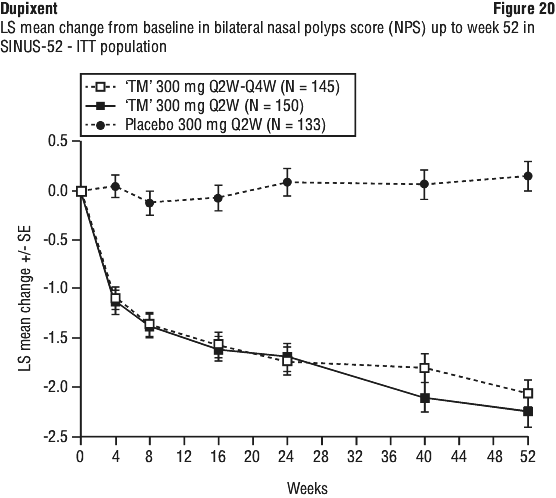 A significant decrease in LMK sinus CT scan score was also observed in SINUS-52 study at week 24 with further improvement at week 52 (see Figure 21). Similar results were seen in SINUS-24 study at week 24.
A significant decrease in LMK sinus CT scan score was also observed in SINUS-52 study at week 24 with further improvement at week 52 (see Figure 21). Similar results were seen in SINUS-24 study at week 24. In both studies, significant improvements in NC and daily loss of smell severity were observed as early as the first assessment at Week 4. The LS mean difference for NC at Week 4 in the Dupixent group versus placebo was 0.41 (95% CI: 0.52, 0.30) in SINUS-24 and -0.37 (95% CI: -0.46, -0.27) in SINUS-52. The LS mean difference for loss of smell at Week 4 in the Dupixent group versus placebo was 0.34 (95% CI: -0.44, -0.25) in SINUS-24 and -0.31 (95% CI: -0.41, -0.22) in SINUS-52.
In both studies, significant improvements in NC and daily loss of smell severity were observed as early as the first assessment at Week 4. The LS mean difference for NC at Week 4 in the Dupixent group versus placebo was 0.41 (95% CI: 0.52, 0.30) in SINUS-24 and -0.37 (95% CI: -0.46, -0.27) in SINUS-52. The LS mean difference for loss of smell at Week 4 in the Dupixent group versus placebo was 0.34 (95% CI: -0.44, -0.25) in SINUS-24 and -0.31 (95% CI: -0.41, -0.22) in SINUS-52. In patients with co-morbid asthma, significant improvement in pre-bronchodilator FEV1 were observed at Week 24 in the pre-specified multiplicity-adjusted pool of the two studies irrespective of baseline blood eosinophils levels. The LS Mean change from baseline in FEV1 at Week 24 for Dupixent 300 mg Q2W was 0.14 vs -0.07 L for placebo, for a difference of 0.21 L (95% CI: 0.13, 0.29).
In patients with co-morbid asthma, significant improvement in pre-bronchodilator FEV1 were observed at Week 24 in the pre-specified multiplicity-adjusted pool of the two studies irrespective of baseline blood eosinophils levels. The LS Mean change from baseline in FEV1 at Week 24 for Dupixent 300 mg Q2W was 0.14 vs -0.07 L for placebo, for a difference of 0.21 L (95% CI: 0.13, 0.29). Improvements in ACQ-6 in patients with co-morbid asthma were observed in both studies. A response was defined as an improvement in score of 0.5 or more. In SINUS-24, at Week 24, the LS mean difference in the Dupixent group versus placebo was 0.76 (95% CI: 1.00 to 0.51).432. In SINUS-52, at Week 52, the LS mean difference in the Dupixent group versus placebo was 0.94 (95% CI: 1.19, 0.69).
Improvements in ACQ-6 in patients with co-morbid asthma were observed in both studies. A response was defined as an improvement in score of 0.5 or more. In SINUS-24, at Week 24, the LS mean difference in the Dupixent group versus placebo was 0.76 (95% CI: 1.00 to 0.51).432. In SINUS-52, at Week 52, the LS mean difference in the Dupixent group versus placebo was 0.94 (95% CI: 1.19, 0.69).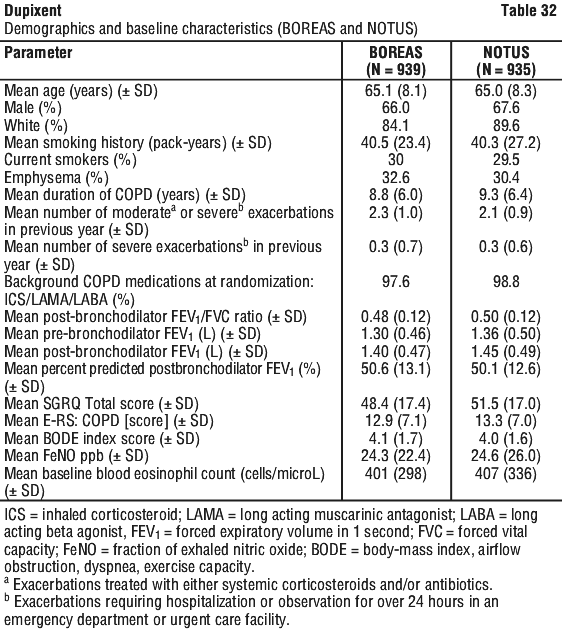
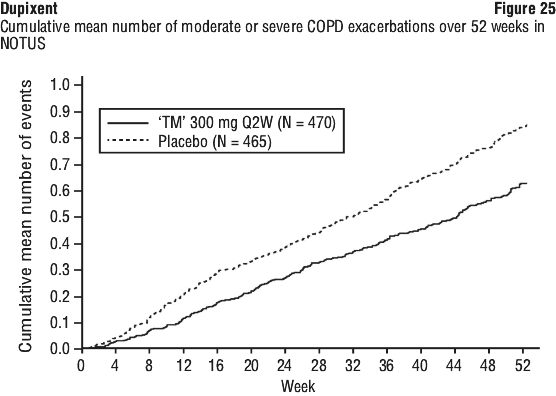
 In both trials, the cumulative mean number of moderate or severe exacerbations observed over 52 weeks was lower in subjects receiving Dupixent compared to placebo (see Figure 24 and Figure 25).
In both trials, the cumulative mean number of moderate or severe exacerbations observed over 52 weeks was lower in subjects receiving Dupixent compared to placebo (see Figure 24 and Figure 25).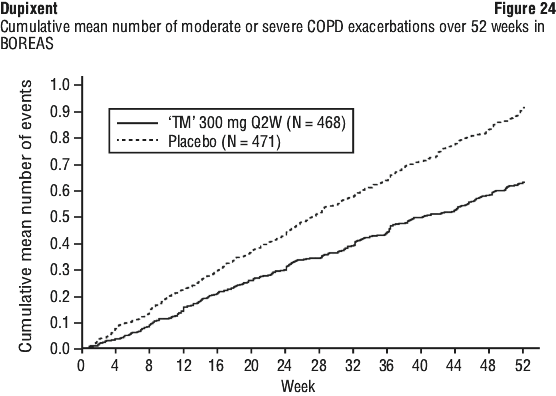
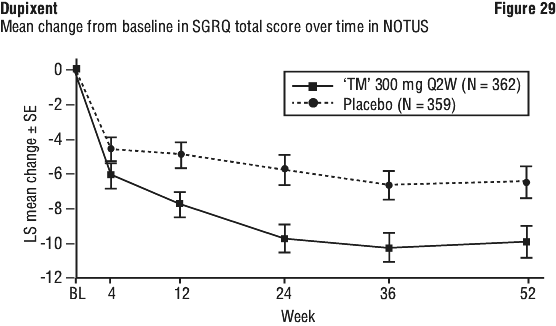
 The time to first moderate or severe COPD exacerbation was longer for patients receiving Dupixent compared to placebo in BOREAS (HR: 0.80; 95% CI: 0.66, 0.98) and NOTUS (HR: 0.71.; 95% CI: 0.57, 0.889).
The time to first moderate or severe COPD exacerbation was longer for patients receiving Dupixent compared to placebo in BOREAS (HR: 0.80; 95% CI: 0.66, 0.98) and NOTUS (HR: 0.71.; 95% CI: 0.57, 0.889).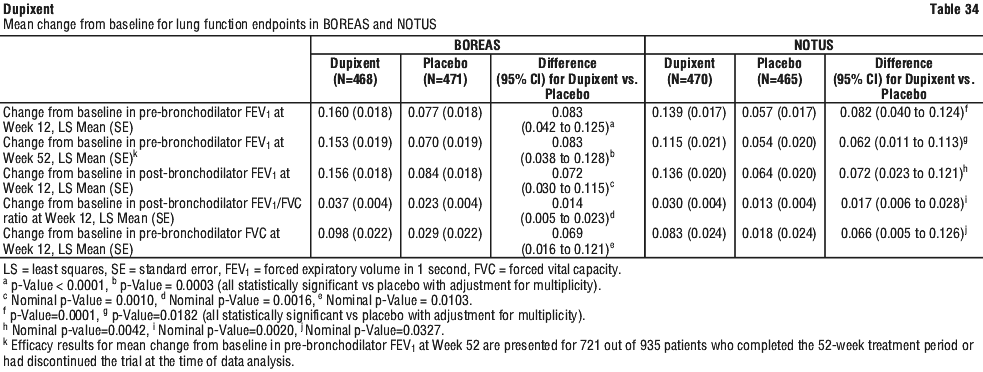
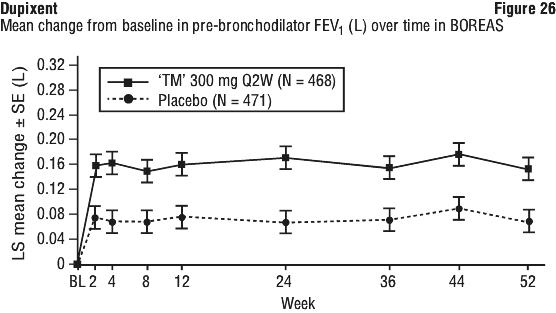
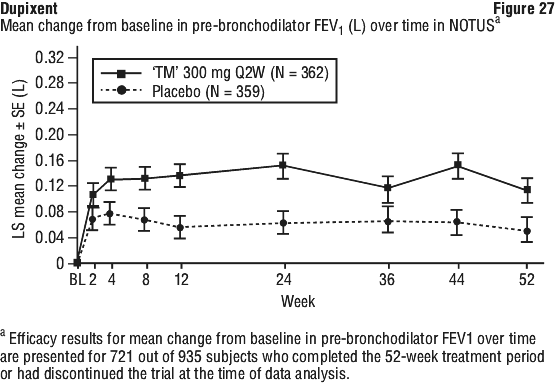 In the subgroup analysis of patients with higher baseline FeNO (≥ 20 ppb) in BOREAS (N=383), treatment with Dupixent statistically significantly improved pre-bronchodilator FEV1 from baseline at Week 12 (LS mean change: 0.232 Dupixent vs 0.108 placebo; LS mean difference: 0.124 [95% CI: 0.045, 0.203]; p=0.002) and Week 52 (LS mean change: 0.247 Dupixent vs 0.120 placebo; LS mean difference: 0.127 [95% CI: 0.042, 0.212]; p=0.003) compared to placebo. In NOTUS, statistically significant improvement from baseline was observed in the subgroup of patients with a higher baseline FeNO (≥ 20 ppb) treated with Dupixent compared to placebo at Week 12 (N=355; LS mean change: 0.221 Dupixent vs 0.081 placebo; LS mean difference: 0.141 [95% CI: 0.058, 0.223]; p=0.001). Treatment with Dupixent improved pre-bronchodilator FEV1 at Week 52 in the subgroup of patients with higher baseline FeNO (≥ 20 ppb) compared to placebo in NOTUS (N=264; LS mean change: 0.176 Dupixent vs 0.095 placebo; LS mean difference: 0.081[95% CI: -0.019, 0.181]) but did not meet statistical significance.
In the subgroup analysis of patients with higher baseline FeNO (≥ 20 ppb) in BOREAS (N=383), treatment with Dupixent statistically significantly improved pre-bronchodilator FEV1 from baseline at Week 12 (LS mean change: 0.232 Dupixent vs 0.108 placebo; LS mean difference: 0.124 [95% CI: 0.045, 0.203]; p=0.002) and Week 52 (LS mean change: 0.247 Dupixent vs 0.120 placebo; LS mean difference: 0.127 [95% CI: 0.042, 0.212]; p=0.003) compared to placebo. In NOTUS, statistically significant improvement from baseline was observed in the subgroup of patients with a higher baseline FeNO (≥ 20 ppb) treated with Dupixent compared to placebo at Week 12 (N=355; LS mean change: 0.221 Dupixent vs 0.081 placebo; LS mean difference: 0.141 [95% CI: 0.058, 0.223]; p=0.001). Treatment with Dupixent improved pre-bronchodilator FEV1 at Week 52 in the subgroup of patients with higher baseline FeNO (≥ 20 ppb) compared to placebo in NOTUS (N=264; LS mean change: 0.176 Dupixent vs 0.095 placebo; LS mean difference: 0.081[95% CI: -0.019, 0.181]) but did not meet statistical significance.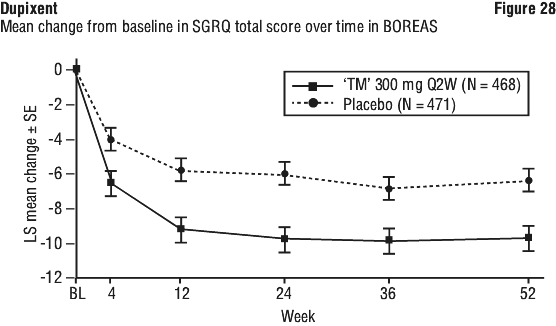
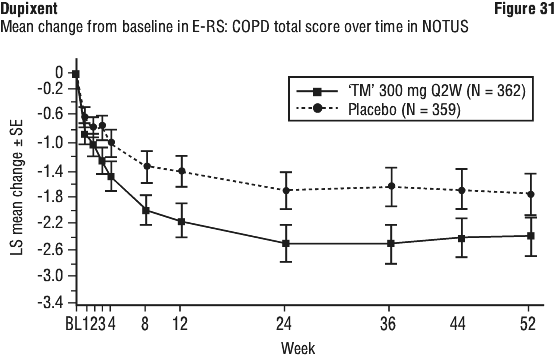
 In BOREAS, Dupixent significantly improved overall respiratory symptoms compared to placebo as measured by LS mean change from baseline in the E-RS:COPD total score at Week 52 (LS mean change: -2.69 Dupixent vs -1.56 placebo; LS mean difference: -1.14 [95% CI: -1.82, -0.45]; p=0.001). Greater improvements in overall respiratory symptoms in patients treated with Dupixent compared to placebo as measured by average weekly change from baseline in the E-RS:COPD total score were observed as early as Week 1 and were maintained through Week 52 (see Figure 30). Consistent improvements were observed across all individual E-RS: COPD domains (cough and sputum, breathlessness, and chest-related symptoms).
In BOREAS, Dupixent significantly improved overall respiratory symptoms compared to placebo as measured by LS mean change from baseline in the E-RS:COPD total score at Week 52 (LS mean change: -2.69 Dupixent vs -1.56 placebo; LS mean difference: -1.14 [95% CI: -1.82, -0.45]; p=0.001). Greater improvements in overall respiratory symptoms in patients treated with Dupixent compared to placebo as measured by average weekly change from baseline in the E-RS:COPD total score were observed as early as Week 1 and were maintained through Week 52 (see Figure 30). Consistent improvements were observed across all individual E-RS: COPD domains (cough and sputum, breathlessness, and chest-related symptoms).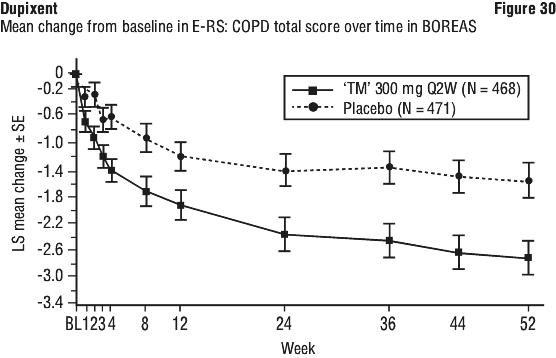
 Dupilumab is a covalent heterotetramer consisting of two disulfide-linked human heavy chains, each covalently linked through a disulfide bond to a human kappa light chain. There is a single N-linked glycosylation site in each heavy chain, located within the CH2 domain of the Fc constant region of the molecule. The dupilumab heavy chain has an immunoglobulin (Ig) G4P isotype constant region. IgG4P is an IgG4 constant region with a single amino acid substitution in the hinge region that recreates the IgG1 hinge sequence in order to stabilise IgG4 dimer formation. The variable domains of the heavy and light chains combine to form the IL-4Rα binding site within the antibody. Dupilumab has a molecular weight of approximately 147 kDa.
Dupilumab is a covalent heterotetramer consisting of two disulfide-linked human heavy chains, each covalently linked through a disulfide bond to a human kappa light chain. There is a single N-linked glycosylation site in each heavy chain, located within the CH2 domain of the Fc constant region of the molecule. The dupilumab heavy chain has an immunoglobulin (Ig) G4P isotype constant region. IgG4P is an IgG4 constant region with a single amino acid substitution in the hinge region that recreates the IgG1 hinge sequence in order to stabilise IgG4 dimer formation. The variable domains of the heavy and light chains combine to form the IL-4Rα binding site within the antibody. Dupilumab has a molecular weight of approximately 147 kDa.