What is in this leaflet
This leaflet answers some common questions about Edronax.
It does not contain all of the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Edronax against the benefits it is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What EDRONAX is used for
What it does
Edronax is used to treat depression.
How it works
Depression is longer lasting and/or more severe than the 'low moods' everyone has from time to time due to the stress of everyday life. It is thought to be caused by a chemical imbalance in parts of the brain.
Edronax may correct this chemical imbalance and help to relieve the symptoms of depression.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
There is no evidence that Edronax is addictive.
This medicine is available only with a doctor's prescription.
Use in Children
There is not enough information to recommend the use of this medicine in children or adolescents younger than 18 years.
Before you take EDRONAX
When you must not take it
Do not take Edronax if you have an allergy to:
- any medicine containing reboxetine
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction to Edronax may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take Edronax if you are taking other medicines used to treat depression such as monoamine oxidase inhibitors (MAOIs).
Do not take Edronax if you have glaucoma (high pressure in the eye).
Do not give this medicine to children or adolescents under the age of 18 years. Safety and effectiveness in children or adolescents younger than 18 years have not been established.
Do not take this medicine after the expiry date (EXP) printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor or pharmacist.
Before you start to take it
Tell your doctor or pharmacist if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor or pharmacist if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor or pharmacist can discuss with you the risks and benefits involved.
Tell your doctor or pharmacist if you have or have had any of the following medical conditions:
- liver or kidney disease
- fits or seizures
- severe mood swings
- difficulty in passing urine
- glaucoma
- prostate disease
- heart problems, including high blood pressure, particularly if you are over 65 years old, heart failure, recent heart attack
- overactive thyroid gland.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines including:
- all prescription medicines
- all medicines, vitamins, herbal supplements or natural therapies you buy without a prescription from a pharmacy, supermarket, naturopath or health food shop.
Some medicines may be affected by Edronax or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines. Your doctor will advise you.
Tell your doctor or pharmacist if you are taking any of the following:
- medicines used to lower blood pressure
- medicines used to treat fungal or bacterial infections
- carbamazepine and phenobarbital, medicines used to control fits or seizures
- potassium-depleting diuretics, medicines used to remove fluid
- fluvoxamine, a medicine used to treat depression
- ergot derivatives or triptans medicines used to treat migraine
- other medicines used to treat mental conditions or pain conditions (e.g., selective serotonin reuptake inhibitors, other serotonin-norepinephrine reuptake inhibitors, tricyclic and tetracyclic antidepressants, lithium, opioids, tryptophan, buspirone, monoamine oxidase inhibitors, and St. John’s Wort).
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take EDRONAX
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how many tablets you need to take each day. This may depend on your age, your condition and whether or not you are taking any other medicines.
The usual dose of Edronax in adults is 4 mg taken twice a day. After 3 weeks your doctor may increase the dose up to 10 mg per day, if required.
If you are over 65 years or have poor kidney or liver function, the usual starting dose is 2 mg taken twice a day. After 3 weeks your doctor may increase the dose up to 6 mg per day, if required.
How to take it
Swallow the tablets whole with a glass of water. Edronax tablets can be broken in half along the breakline if your doctor has prescribed a half tablet.
When to take it
Take your medicine at about the same time each day, once in the morning and once at night. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food.
How long to take it
Most antidepressants take some time to work, so do not be discouraged if you do not feel better straight away. Some of your symptoms may improve after two weeks but it may take four to six weeks for you to start to feel the full benefit of Edronax. You may need to take Edronax for several months or longer.
Continue taking your medicine for as long as your doctor tells you. This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Australian Poisons Information Centre (telephone 13 11 26) or the New Zealand National Poisons Information Centre (telephone 0800 POISON or 0800 764 766), or go to Accident and Emergency at your nearest hospital, if you think that you or anyone else may have taken too much Edronax.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you take too much Edronax you may feel nervous, dizzy or have a fast heart rate.
While you are taking EDRONAX
Things you must do
If you become pregnant while taking this medicine, tell your doctor or pharmacist immediately.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Edronax.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
Tell your doctor if, for any reason, you have not taken your tablets exactly as prescribed. Otherwise your doctor may think that it was not effective and may change your treatment unnecessarily.
Tell your doctor immediately if you have any suicidal thoughts or other mental/mood changes. A worsening of depressive symptoms including thoughts of suicide or self-harm may occur in the first one or two months of you taking Edronax or when the doctor changes your dose. These symptoms should be controlled when the full effect of Edronax takes place.
Young adults under 24 years of age are more likely to experience these effects during the first few months of treatment.
Patients and caregivers should be alert and monitor for these effects.
Signs and symptoms of suicide include:
- Thoughts or talk of death or suicide
- Thoughts or talk of self-harm or harm to others
- Any recent attempts of self-harm
- Increase in aggressive behaviour, irritability or agitation.
Any mention of suicide or violence must be taken seriously.
If you or someone you know is demonstrating these warning signs of suicide while taking Edronax, contact your doctor or a mental health professional right away.
Discuss with your doctor any problems you may have and how you feel, especially any feelings of severe sadness or unusual bursts of energy or anger. This will help the doctor determine the best treatment for you.
Keep all your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not take Edronax to treat any other complaints unless your doctor or pharmacist tells you to.
Do not take any other medicines, whether they require a prescription or not, without first telling your doctor.
Do not stop taking your medicine, or change the dosage, without checking with your doctor or pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how Edronax affects you. As with other medicines for depression, this medicine may cause dizziness or drowsiness in some people. Make sure you know how you react to Edronax before you drive a car, operate machinery, or do anything else that could be dangerous.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Edronax. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
It can be difficult to tell whether side effects are the result of taking Edronax, effects of your condition or side effects of other medicines you may be taking. For this reason it is important to tell your doctor of any change in your condition.
Do not be alarmed by the list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if...
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- dry mouth
- headache
- nausea, vomiting
- constipation or diarrhoea
- difficulty sleeping
- increased sweating
- fast heart beat
- dizziness
- eye problems
- difficulty passing urine
- sexual problems
- stomach pain
- unusual tiredness or weakness
- tingling or numbness of the hands or feet.
- decreased appetite
- altered taste
- restless leg syndrome
- hair and skin problems
- flushing of the skin (sudden reddening of the face, neck, or upper chest) due to increased blood flow
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor as soon as possible if...
Tell your doctor as soon as possible if you notice any of the following:
- anxiety, agitation
- confusion
- hallucinations, delirium
- bleeding or bruising more easily than normal
- frequent signs of infection such as fever, sore throat, severe chills, mouth ulcers
- coldness or loss of blood to your extremities
- involuntary muscle movements such as tremor, clonus (muscle contractions or spasms that lead to muscle tightness and pain), and hyperreflexia (overactive reflexes)
- tiredness, headache, shortness of breath when exercising, dizziness or pale skin.
Go to hospital if...
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- fits or seizures
- chest pain
- thoughts of suicide or attempting suicide or self harm
- aggressive behaviour
- change in heart beat (fast, slow, irregular), sometimes with fainting/loss of consciousness
- itching skin rash or hives
- shortness of breath, wheezing or trouble breathing
- swelling of the face, lips, tongue or other part of the body.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation. These side effects are very rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell. Other side effects not listed above may occur in some people.
Some of these side effects (for example, high cholesterol, abnormal liver function tests) can only be found when your doctor does tests from time to time to check your progress.
After taking EDRONAX
Storage
Keep your tablets in the pack or bottle until it is time to take them. If you take the tablets out of the pack or bottle they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 25°C.
Do not store Edronax or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor or pharmacist tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
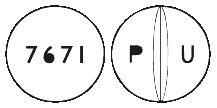
Edronax Tablets 4 mg are white, convex, round tablets with a breakline on one side and engraved 'P' on the left of the breakline and 'U' on the right side of the breakline, and '7671' on the opposite side.
Ingredients
Active ingredient
Reboxetine mesilate.
Each 4 mg tablet contains 4 mg of reboxetine as mesilate.
Other ingredients:
- magnesium stearate
- microcrystalline cellulose
- silicon dioxide
- crospovidone
- calcium hydrogen phosphate dihydrate.
This medicine does not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
Supplier
Edronax is supplied in Australia by:
Pfizer Australia Pty Ltd
Sydney NSW 2000
Toll Free number: 1800 675 229
www.pfizermedinfo.com.au
Australian Registration Number
4 mg tablets: AUST R 79745
This leaflet was prepared in November 2022.
® Registered trademark.
Published by MIMS January 2023

 There was an increase in heart rate upon standing to values ≥ 100 beats/min mainly in adult patients (20% of the patients on short-term treatment compared with 6% on placebo, and 23% of the patients on long-term treatment compared with 17% on placebo). In all short-term controlled studies in depression, the mean change in pulse (in beats per minute) for reboxetine treated patients was 2.9, 8.3 and 3.0 in the supine, sitting and standing positions, respectively, compared with -0.5, 0 and 0 for placebo treated patients in the corresponding positions.
There was an increase in heart rate upon standing to values ≥ 100 beats/min mainly in adult patients (20% of the patients on short-term treatment compared with 6% on placebo, and 23% of the patients on long-term treatment compared with 17% on placebo). In all short-term controlled studies in depression, the mean change in pulse (in beats per minute) for reboxetine treated patients was 2.9, 8.3 and 3.0 in the supine, sitting and standing positions, respectively, compared with -0.5, 0 and 0 for placebo treated patients in the corresponding positions. For long-term tolerability, 143 Edronax treated and 140 placebo treated adult patients participated in a long-term placebo controlled study. Adverse events newly emerged on long-term treatment in 28% of the Edronax treated patients and 23% of the placebo treated patients, and caused discontinuation in 4% and 1% of the cases, respectively. There was a similar risk of the development of individual events with Edronax and placebo. Among events seen more than occasionally, no individual events not seen on short-term treatment were apparent.
For long-term tolerability, 143 Edronax treated and 140 placebo treated adult patients participated in a long-term placebo controlled study. Adverse events newly emerged on long-term treatment in 28% of the Edronax treated patients and 23% of the placebo treated patients, and caused discontinuation in 4% and 1% of the cases, respectively. There was a similar risk of the development of individual events with Edronax and placebo. Among events seen more than occasionally, no individual events not seen on short-term treatment were apparent.
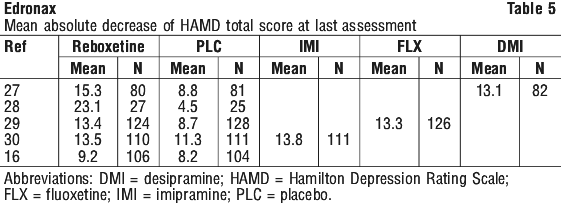 The mean HAMD total score improvement associated with Edronax treatment ranged from 9.2 to 23.1 points. The mean HAMD improvement associated with placebo was always lower than that associated with Edronax, with an average decrease of 4.5 to 11.3 points. The difference between Edronax and placebo was statistically significant in three of the five studies above. In two studies, the difference between Edronax and placebo was not statistically significant on the mean HAMD total score. In one of these studies, the active comparator imipramine also failed to show a statistically significant difference over placebo. In the other study, a number of secondary efficacy measures demonstrated statistically significant differences in favour of Edronax over placebo.
The mean HAMD total score improvement associated with Edronax treatment ranged from 9.2 to 23.1 points. The mean HAMD improvement associated with placebo was always lower than that associated with Edronax, with an average decrease of 4.5 to 11.3 points. The difference between Edronax and placebo was statistically significant in three of the five studies above. In two studies, the difference between Edronax and placebo was not statistically significant on the mean HAMD total score. In one of these studies, the active comparator imipramine also failed to show a statistically significant difference over placebo. In the other study, a number of secondary efficacy measures demonstrated statistically significant differences in favour of Edronax over placebo.


