
What is in this leaflet
This leaflet answers some common questions about Eliquis.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Eliquis against the benefits it is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Eliquis is used for
What Eliquis does
This medicine is used to:
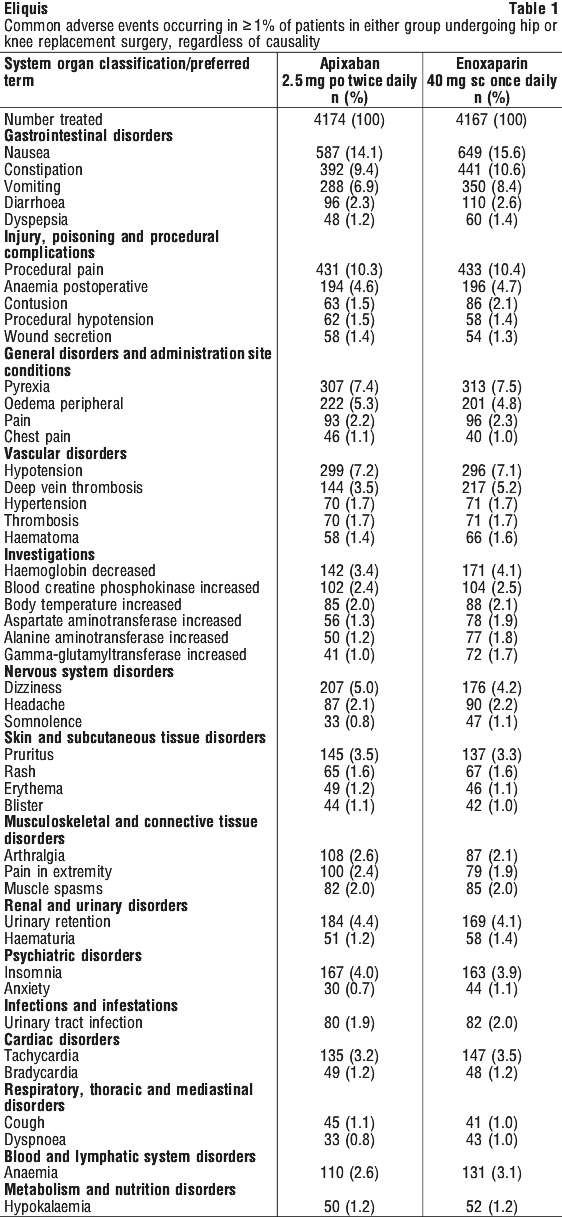
- prevent blood clots in your veins after a hip or knee replacement surgery.
After an operation you are at an increased risk of getting blood clots. - treat blood clots occurring in deep veins (Deep Vein Thrombosis [DVT]) and clots in your lungs (Pulmonary Embolism [PE]) and also to prevent these from recurring.
- prevent stroke and blood clots in a condition called atrial fibrillation, which is a type of abnormal heart rhythm.
With atrial fibrillation, part of the heart does not beat the way it should. This can lead to blood clots forming and increase your risk of having a stroke.
How Eliquis works
The active substance in Eliquis is apixaban. It belongs to a group of medicines called antithrombotic agents.
It works by inhibiting a blood clot forming substance called Factor Xa.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
This medicine is not expected to affect your ability to drive a car or operate machinery.
Use in Children
There is not enough information to recommend the use of this medicine in children and adolescents under the age of 18 years.
Before you take Eliquis
When you must not take it
Do not take Eliquis if you have an allergy to:
- any medicine containing apixaban
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take Eliquis if you have, or have had, any of the following medical conditions:
- any disease or injury to a body organ that is actively bleeding or at high risk of bleeding e.g. bleeding ulcer in the stomach or bowel, recent bleeding in the brain, cancer at high risk of bleeding
- liver disease which leads to an increased risk of bleeding
- severely reduced kidney function. Your doctor will determine your kidney function
- a recent operation on the brain, spinal column or eye(s)
- recent brain or spine injury
- abnormalities of any blood vessels that may lead to an increase in bleeding
- any blood vessel abnormalities of your oesophagus or "gullet"
- any disease or injury to a body organ that could lead to significant bleeding e.g. stomach ulcers, bowel ulcers.
Do not take Eliquis if you are taking the following medicines:
- medicines for fungal infections e.g. ketoconazole, itraconazole, voriconazole or posaconazole, unless they are only applied to the skin
- anti-viral medicines for HIV/AIDS e.g. ritonavir
- other medicines to stop your blood from clotting e.g. heparin, enoxaparin, warfarin, rivaroxaban or dabigatran.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have ever been hospitalised for a heart condition (heart attack or unstable angina).
Tell your doctor if you have ever received a stent placed in the coronary arteries of your heart.
Tell your doctor if you are 75 years or older or if you have or have had any of the following medical conditions which may lead to an increased risk of bleeding:
- a heart condition known as bacterial endocarditis
- type of stroke called "haemorrhagic stroke"
- blood disorders that affect your ability to form clots and stop bleeding
- recent or past ulcer of your stomach or bowel
- moderate or mild kidney disease
- liver disease
- have a lung condition called bronchiectasis
- have had a history of bleeding in your lungs
- high blood pressure that is not controlled with medications.
Your doctor may decide to keep you under closer observation.
Tell your doctor if you have a prosthetic heart valve or severe rheumatic heart disease, especially mitral stenosis (problem with the mitral valve in your heart).
Tell your doctor if you know that you have a disease called antiphospholipid syndrome (a disorder of the immune system that causes an increased risk of blood clots). Your doctor who will decide if the treatment may need to be changed.
If you are having hip or knee replacement surgery and your operation involves a catheter or injection into your spinal column e.g. for epidural or spinal anaesthesia or pain reduction:
- tell your doctor immediately if you get numbness or weakness of your legs or problems with your bowel or bladder after the end of anaesthesia, because urgent care is necessary.
Tell your doctor if you are pregnant or plan to become pregnant. Eliquis is not recommended for use in pregnant women. Your doctor can discuss with you the risks and benefits involved.
Tell your doctor if you are breast-feeding or intend to breast-feed. Eliquis is not recommended during breast-feeding. You should not breast-feed and take Eliquis at the same time. The active ingredient in Eliquis may be present in breast milk and poses a bleeding risk to the baby. You and your doctor should decide if you will take Eliquis or breast-feed.
If you have not told your doctor about any of the above, tell him/her before you start taking Eliquis.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including:
- all prescription medicines
- all medicines, vitamins, herbal supplements or natural therapies you buy without a prescription from a pharmacy, supermarket, naturopath or health food shop.
Some medicines may be affected by Eliquis or may affect how it works. You may need different amounts of your medicines, or you may need to take different medicines. Your doctor will advise you.
Tell your doctor or pharmacist if you are taking any of the following:
- medicines used to treat fungal infections such as ketoconazole (Nizoral®), itraconazole (Sporanox®), voriconazole (Vfend®) and posaconazole (Noxafil®)
- anti-viral medicines for HIV/AIDS e.g. ritonavir (Norvir®)
- rifampin or rifampicin (Rifadin®)
- medicines to treat epilepsy such as phenytoin (Dilantin®), carbamazepine (Tegretol®), phenobarbitone
- St John's wort
- medicines to treat depression such as sertraline, citalopram and venlafaxine
- medicines used to treat pain and inflammation including non-steroidal anti-inflammatory drugs (NSAIDs) such as naproxen (Naprosyn®) or aspirin (Aspro®)
- other medicines used to prevent blood clots such as enoxaparin (Clexane®), clopidogrel (Iscover®, Plavix®), ticagrelor (Brilinta), prasugrel (Effient), heparin, fondaparinux (Arixtra®), bivalirudin (Angiomax®), rivaroxaban (Xarelto®), dabigatran (Pradaxa®), dipyridamole (Persantin®)
- quinidine
- verapamil
- diltiazem
- amiodarone
- the antibiotic, clarithromycin.
(Not all brand names are given above so check with your doctor or pharmacist).
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take Eliquis
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how many tablets you need to take each day.
If you are having hip or knee replacement surgery
The recommended dose is one 2.5 mg tablet taken twice a day.
If you have blood clots
The recommended dose to treat blood clots is two 5 mg tablets taken twice a day for 7 days, then one 5 mg tablet taken twice a day.
The recommended dose to prevent blood clots that recur is one 2.5 mg tablet taken twice daily.
If you have atrial fibrillation
The recommended dose is normally one 5 mg tablet taken twice a day.
The recommended dose is one 2.5 mg tablet taken twice a day if you meet any two of the following:
- are 80 years or older
- weigh 60 kilograms or under
- have reduced kidney function.
How to take it
Swallow the tablet(s) whole with a full glass of water.
If you are unable to swallow the tablet(s) whole, follow the steps below to crush the tablet(s). This will help make sure that all of the crushed tablet(s) will be taken.
- use a mortar and pestle or a similar device to crush the tablet(s)
- transfer the powder to a drinking glass or a small bowl
- when using water, 5% dextrose in water or apple juice:
- add a small amount of water/5% dextrose in water/apple juice (30 mL) to the mortar and pestle/device and stir
- transfer the water/5% dextrose in water/apple juice to the drinking glass
- mix the powder with the water/5% dextrose in water/apple juice and drink right away
- rinse the glass with a small amount of water/5% dextrose in water/apple juice and drink right away. - when using apple sauce:
- mix the powder with a small amount of apple sauce (30 g) in a small bowl and eat with a spoon right away
- add a small amount of water (30 mL) to the mortar and pestle/device and stir
- transfer the water to the bowl and drink right away
- rinse the bowl and the spoon with a small amount of water and drink right away.
If necessary, your doctor may also give you the crushed tablet(s) mixed in 60 mL of water or 5% dextrose in water, through a nasogastric tube.
It does not matter if you take this medicine before or after food.
When to take it
Take the first tablet as directed by your doctor.
If you are having hip or knee replacement surgery it is usual to start taking your tablets 12 to 24 hours after your operation.
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
If you stop taking it suddenly, you may be at an increased risk of developing a blood clot, which can lead to serious problems such as a stroke if you have atrial fibrillation.
If you are having hip or knee replacement surgery
If you have had a hip replacement, you will usually take the tablets for about 5 weeks.
If you have had a knee replacement, you will usually take the tablets for about 2 weeks.
If you have blood clots
To treat blood clots you will usually take the tablets for up to 6 months. If necessary, you may need to continue taking the tablets, usually at a lower dose, to prevent further blood clots.
If you have atrial fibrillation
Continue taking your medicine for as long as your doctor tells you.
If you require cardioversion
If your abnormal heartbeat needs to be restored by a procedure called cardioversion, take your medicine at the times your doctor tells you, to help prevent blood clots that may result in strokes or other problems.
If you forget to take it
Take your next tablet as soon as you remember, then continue taking the tablets as normal (twice a day).
Do not take a double dose to make up for the forgotten tablet.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor, or the Poisons Information Centre on 13 11 26 for advice, or go to the Accident and Emergency Department at the nearest hospital, if you think that you or anyone else may have taken too much Eliquis.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Symptoms of an overdose may include bleeding that does not stop.
While you are taking Eliquis
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Eliquis.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
If you are going to have any surgery or procedure, including dental surgery, tell your surgeon, doctor or dentist that you are taking this medicine.
Eliquis should be temporarily stopped before surgery.
Your doctor will tell you when to stop using Eliquis before your surgery or procedure.
Your doctor will also tell you when you can start taking Eliquis after your surgery.
Tell your doctor that you are taking Eliquis if your doctor is planning for you to have an anaesthetic injection in your back (spinal or epidural injection).
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not take Eliquis to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without first checking with your doctor. If you stop taking it suddenly, your condition may worsen or you may have serious side effects.
Things to be careful of
Eliquis contains lactose. If you have been told by your doctor that you have an intolerance to some sugars, contact your doctor before taking it.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Eliquis.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
It can be difficult to tell whether side effects are the result of taking Eliquis, effects of your condition or side effects of other medicines you may be taking. For this reason it is important to tell your doctor of any change in your condition.
Do not be alarmed by the list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if...
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- tiredness, weakness, paleness, dizziness, light-headedness, headache - which can be due to low iron in the blood
- bleeding from any part of your body, no matter how minor, as these may be difficult to control
- bruising
- nausea (feeling sick), vomiting
- diarrhoea or constipation
- fever
- sore nasal passages and throat
- frequent need to urinate or pain while urinating
- coughing.
The above list includes the more common side effects of your medicine.
Tell your doctor as soon as possible if...
Tell your doctor as soon as possible if you notice any of the following:
- excessive bleeding or prolonged bleeding. There is no antidote to reverse this bleeding. It is important to contact your doctor immediately if you experience excessive or prolonged bleeding.
- stomach swelling, yellowing of the skin or whites of the eyes (due to liver problems)
- oozing from your surgical wound
- swelling of the hands, ankles or feet due to water retention
- blood in urine, reduced urine output, swelling of the legs, ankles and feet, increased time for blood to clot (high INR test values) and heavy bleeding. These symptoms may be related to a condition called anticoagulant-related nephropathy.
The above list includes serious side effects that may require medical attention.
Go to hospital if...
Tell your doctor immediately or go to the Accident and Emergency Department at your nearest hospital, if you notice any of the following:
- bleeding from your nose
- if you have dark brown urine or blood in your urine
- if you cough up blood
- if you vomit and it is black
- if you have black stools or blood in your stools
- excessive bleeding or prolonged bleeding. There is no antidote to reverse this bleeding. It is important to contact your doctor immediately if you experience excessive or prolonged bleeding.
- if you have an allergic reaction to Eliquis. Symptoms may include shortness of breath, wheezing or difficulty breathing, swelling of the face, lips, tongue or other parts of the body, rash, itching or hives on the skin.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some people.
Some of the other possible side effects (for example, changes in liver or kidney function) can only be found when your doctor does tests from time to time to check your progress.
After taking Eliquis
Storage
Keep your tablets in the pack until it is time to take them. If you take the tablets out of the pack they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store Eliquis or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Eliquis 2.5 mg tablets are yellow and round with "893" on one side and "2 1/2" on the other.
They are packed in blister packs in cartons of 10, 14, 20, 30, 60 or 100 tablets.
Eliquis 5 mg tablets are pink and oval-shaped with "894" on one side and "5" on the other.
They are packed in blister packs in cartons of 14, 20, 28, 56, 60, 100, 112, 120 and 168 tablets.
Not all pack sizes may be available.
Ingredients
Eliquis tablets contain 2.5 mg or 5 mg of apixaban as the active ingredient.
They also contain:
- lactose
- lactose monohydrate
- microcrystalline cellulose
- croscarmellose sodium
- sodium lauryl sulfate
- magnesium stearate
- hypromellose
- titanium dioxide
- glycerol triacetate
- yellow iron oxide (2.5 mg tablets)
- red iron oxide (5 mg tablets)
This medicine does not contain sucrose, gluten, tartrazine or any other azo dyes.
Sponsored and Supplied by:
Bristol-Myers Squibb Australia Pty Ltd
4 Nexus Court
Mulgrave VIC 3170
Toll free number: 1800 067 567
[email protected]
Also distributed by:
Pfizer Australia Pty Ltd
Sydney NSW
Toll Free Number: 1800 675 229
Australian registration numbers
2.5 mg tablet: AUST R 172244
5 mg tablet: AUST R 193474
Date of preparation
This leaflet was prepared in May 2023.
® ELIQUIS is a registered trademark of Bristol-Myers Squibb Company.
© Copyright of Bristol-Myers Squibb Company 2019-2023.
Published by MIMS June 2023
 Intracranial haemorrhage was reduced > 50% with apixaban. GUSTO severe and TIMI major bleeding were reduced > 40% with apixaban. Fatal bleeding was reduced > 70% with apixaban.
Intracranial haemorrhage was reduced > 50% with apixaban. GUSTO severe and TIMI major bleeding were reduced > 40% with apixaban. Fatal bleeding was reduced > 70% with apixaban.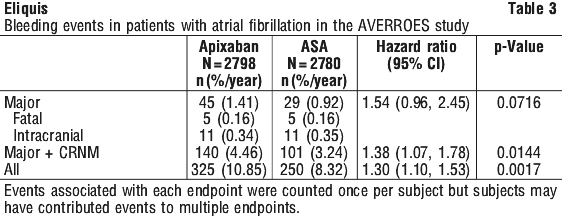 Treatment discontinuation due to bleeding related adverse reactions occurred in 1.5% and 1.3% of patients treated with apixaban and ASA, respectively.
Treatment discontinuation due to bleeding related adverse reactions occurred in 1.5% and 1.3% of patients treated with apixaban and ASA, respectively.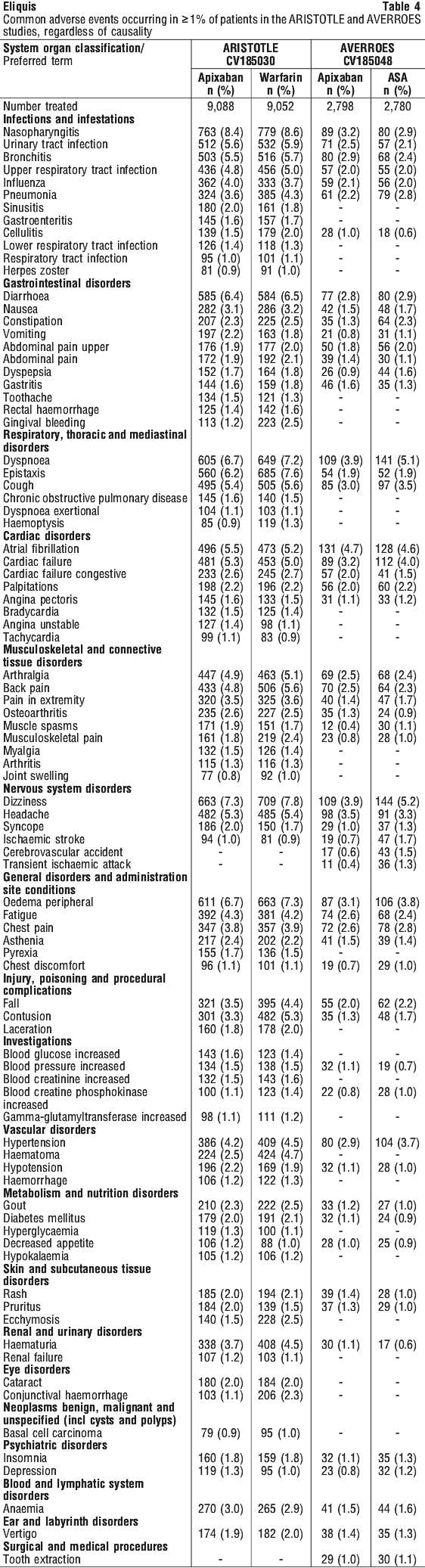 Adverse reactions in the ARISTOTLE and AVERROES studies are listed below by system organ classification (MedDRA) and by frequency. The frequency assignments are primarily based on the frequencies observed in the ARISTOTLE study. The adverse reactions observed in the AVERROES study were consistent with those observed in the ARISTOTLE study.
Adverse reactions in the ARISTOTLE and AVERROES studies are listed below by system organ classification (MedDRA) and by frequency. The frequency assignments are primarily based on the frequencies observed in the ARISTOTLE study. The adverse reactions observed in the AVERROES study were consistent with those observed in the ARISTOTLE study.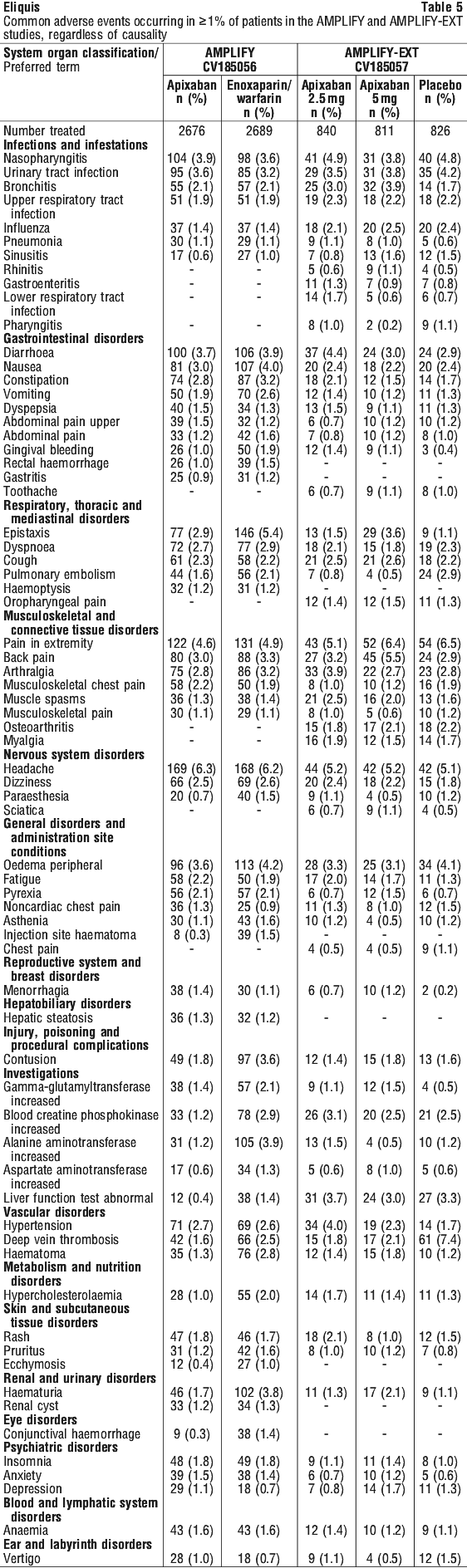 Adverse reactions in the AMPLIFY and AMPLIFY-EXT studies are listed by system organ classification (MedDRA) and by frequency.
Adverse reactions in the AMPLIFY and AMPLIFY-EXT studies are listed by system organ classification (MedDRA) and by frequency.
 Although treatment with apixaban at the recommended dose does not require routine monitoring of exposure, the use of a calibrated quantitative anti-FXa assay may be useful in exceptional situations where knowledge of apixaban exposure may help to inform clinical decisions, e.g. overdose or emergency surgery.
Although treatment with apixaban at the recommended dose does not require routine monitoring of exposure, the use of a calibrated quantitative anti-FXa assay may be useful in exceptional situations where knowledge of apixaban exposure may help to inform clinical decisions, e.g. overdose or emergency surgery.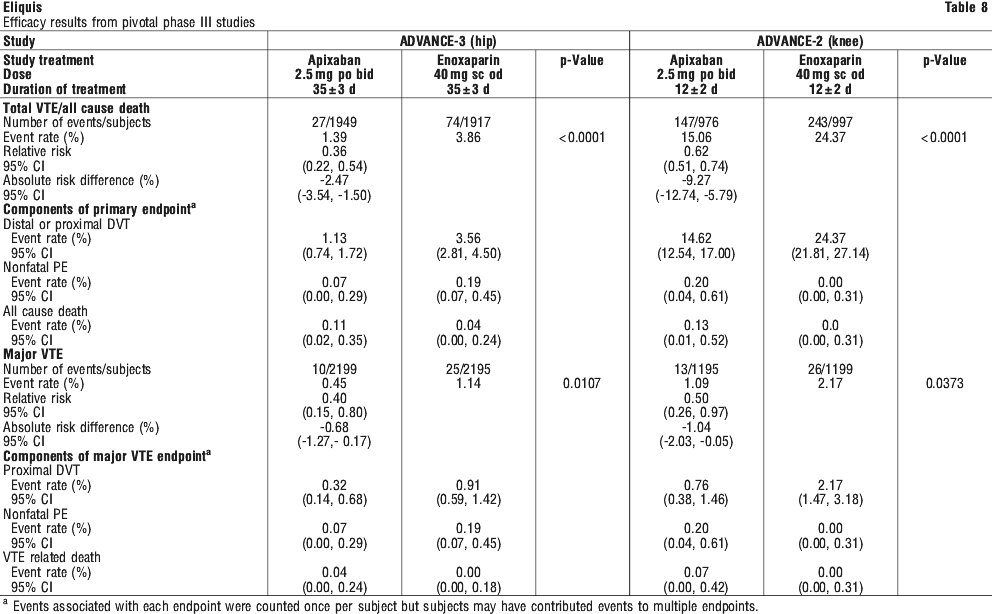 The safety endpoints of major bleeding, the composite of major and clinically relevant nonmajor (CRNM) bleeding, and all bleeding showed similar rates for patients treated with apixaban 2.5 mg compared with enoxaparin 40 mg (see Table 8). Major bleeding was defined as a decrease in haemoglobin of 20 g/L or more over a 24 hour period, transfusion of 2 or more units of packed red cells, bleeding into a critical site (e.g. intracranial haemorrhage) or fatal. CRNM bleeding was defined as significant epistaxis, gastrointestinal bleed, significant haematuria, significant haematoma, bruising or ecchymosis, or haemoptysis. All the bleeding criteria included surgical site bleeding.
The safety endpoints of major bleeding, the composite of major and clinically relevant nonmajor (CRNM) bleeding, and all bleeding showed similar rates for patients treated with apixaban 2.5 mg compared with enoxaparin 40 mg (see Table 8). Major bleeding was defined as a decrease in haemoglobin of 20 g/L or more over a 24 hour period, transfusion of 2 or more units of packed red cells, bleeding into a critical site (e.g. intracranial haemorrhage) or fatal. CRNM bleeding was defined as significant epistaxis, gastrointestinal bleed, significant haematuria, significant haematoma, bruising or ecchymosis, or haemoptysis. All the bleeding criteria included surgical site bleeding.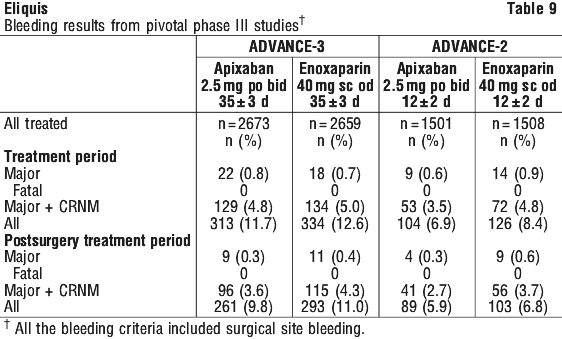
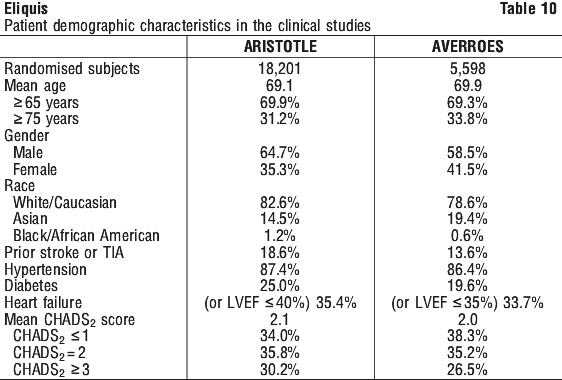
 Centres were ranked post hoc by the percentage of time that warfarin treated patients were in therapeutic range (INR 2-3). Findings for stroke/ systemic embolism, major bleeds, and all cause mortality are shown for centres above and below the median level of INR control in Table 12. The benefits of apixaban relative to warfarin were consistent in patients enrolled at centres with INR control below or above the median.
Centres were ranked post hoc by the percentage of time that warfarin treated patients were in therapeutic range (INR 2-3). Findings for stroke/ systemic embolism, major bleeds, and all cause mortality are shown for centres above and below the median level of INR control in Table 12. The benefits of apixaban relative to warfarin were consistent in patients enrolled at centres with INR control below or above the median.
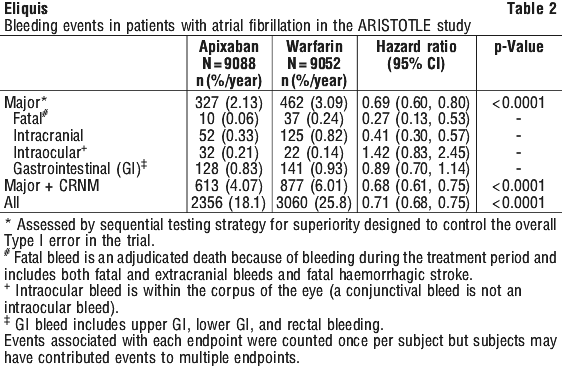
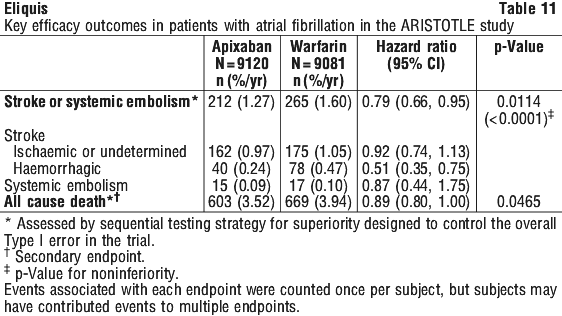
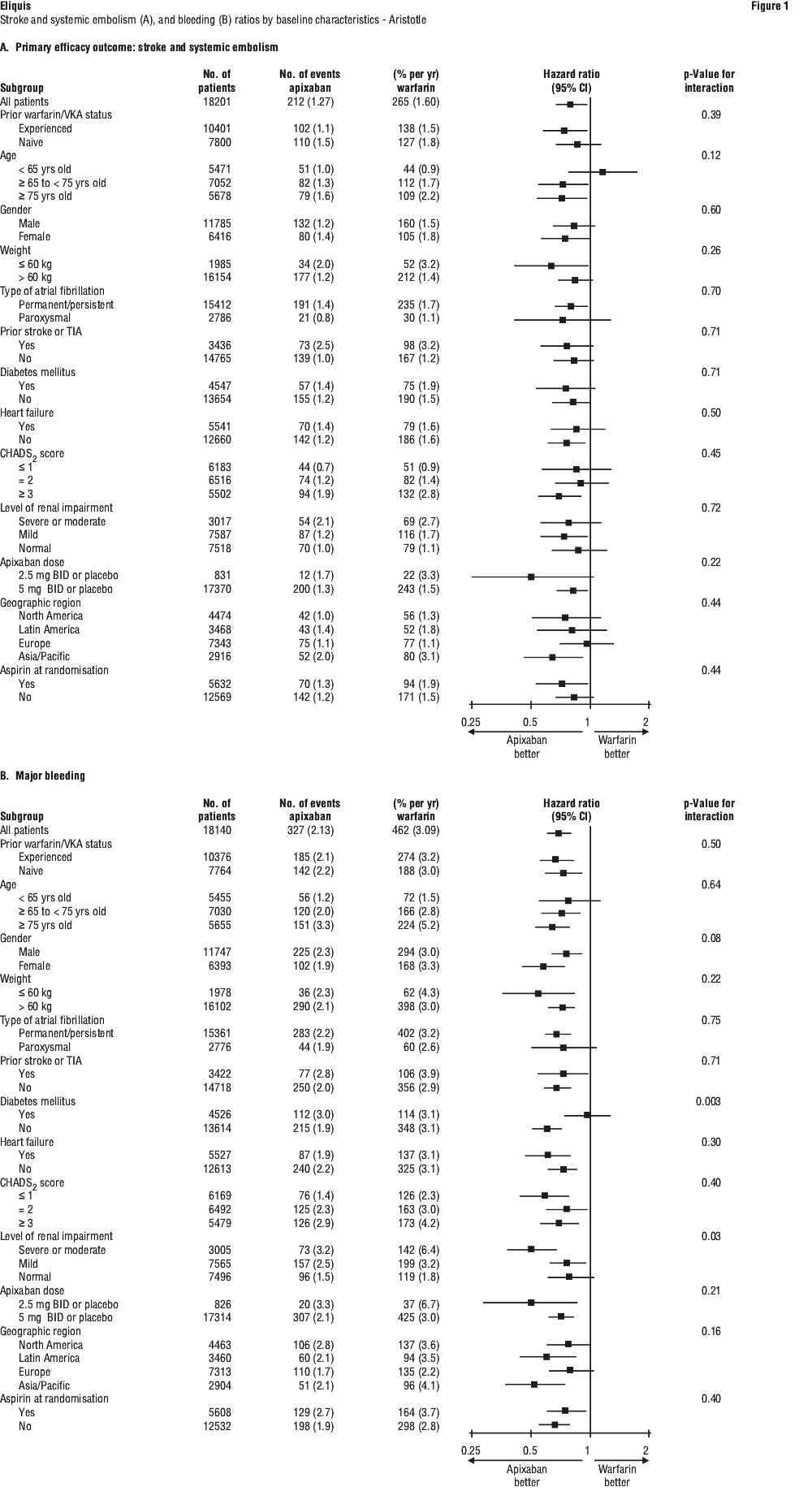 The ARISTOTLE study shows that the risk of stroke, death and major bleeding increases significantly with age. Eliquis compared with warfarin was shown to reduce these outcomes in a consistent manner regardless of age (see Section 4.4, Use in the elderly).
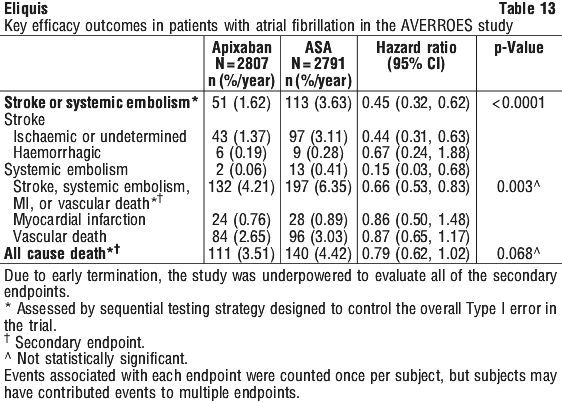
The ARISTOTLE study shows that the risk of stroke, death and major bleeding increases significantly with age. Eliquis compared with warfarin was shown to reduce these outcomes in a consistent manner regardless of age (see Section 4.4, Use in the elderly).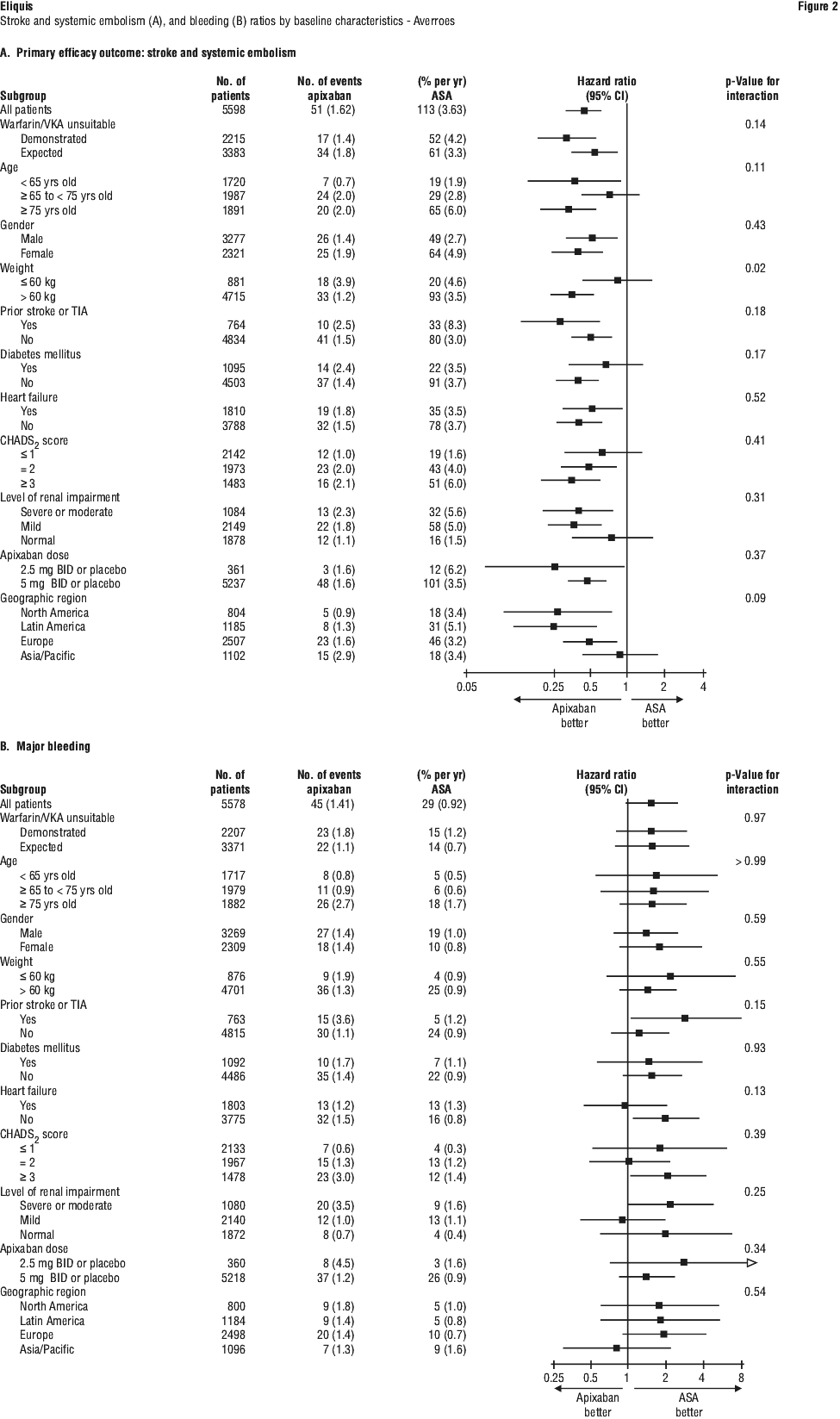 In a clinical study in high risk acute coronary syndrome patients, as characterised by advanced age and multiple cardiac and noncardiac comorbidities (e.g. diabetes, heart failure), receiving apixaban 5 mg twice daily versus placebo, a significant increase in bleeding risk, including gastrointestinal and intracranial bleeding, was reported with the triple combination of apixaban, ASA and clopidogrel (see Section 4.5, Effect of other medicines on apixaban).
In a clinical study in high risk acute coronary syndrome patients, as characterised by advanced age and multiple cardiac and noncardiac comorbidities (e.g. diabetes, heart failure), receiving apixaban 5 mg twice daily versus placebo, a significant increase in bleeding risk, including gastrointestinal and intracranial bleeding, was reported with the triple combination of apixaban, ASA and clopidogrel (see Section 4.5, Effect of other medicines on apixaban).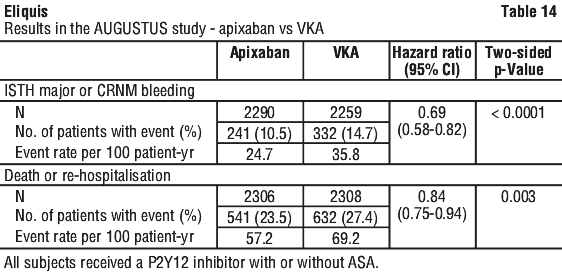 For the ASA versus placebo comparison, the addition of ASA to anticoagulation (with either apixaban or VKA) on top of P2Y12 inhibitor significantly increased the risk of ISTH major or CRNM bleeding (HR = 1.88, 95% CI 1.58, 2.23; two-sided p < 0.0001). A subgroup analysis found that concomitant use of ASA resulted in a near doubling of the risk of major or CRNM bleeding in both apixaban-treated and VKA treated subjects. See Table 15 for results of the primary safety outcome.
For the ASA versus placebo comparison, the addition of ASA to anticoagulation (with either apixaban or VKA) on top of P2Y12 inhibitor significantly increased the risk of ISTH major or CRNM bleeding (HR = 1.88, 95% CI 1.58, 2.23; two-sided p < 0.0001). A subgroup analysis found that concomitant use of ASA resulted in a near doubling of the risk of major or CRNM bleeding in both apixaban-treated and VKA treated subjects. See Table 15 for results of the primary safety outcome.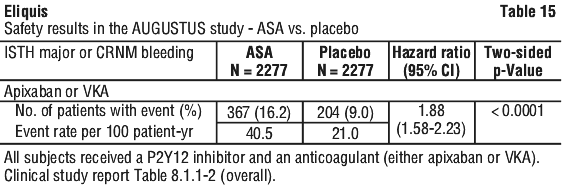
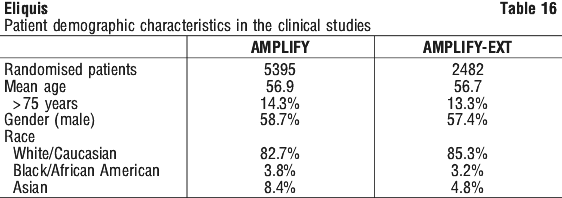
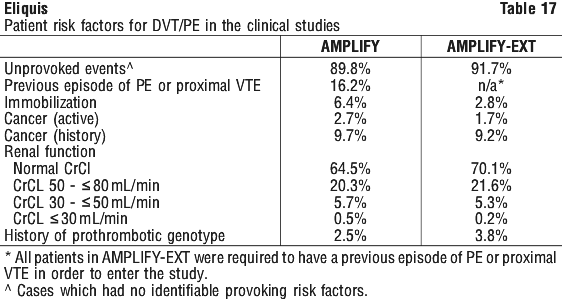
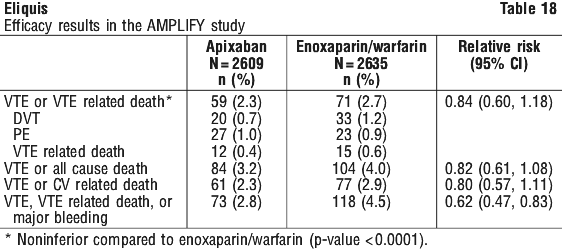 Figure 3 is a plot of the time from randomisation to the occurrence of the first primary efficacy endpoint event in the two treatment groups in the AMPLIFY study.
Figure 3 is a plot of the time from randomisation to the occurrence of the first primary efficacy endpoint event in the two treatment groups in the AMPLIFY study.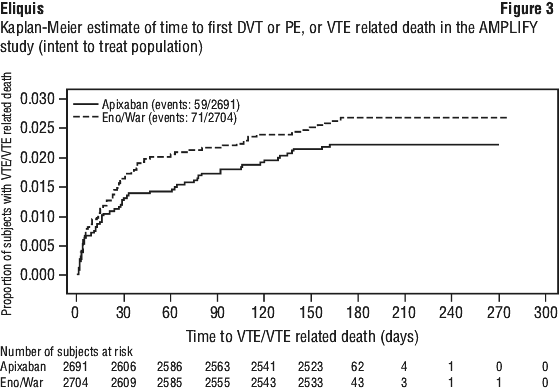 Apixaban efficacy in initial treatment of VTE was consistent between patients who were treated for a PE [Relative Risk 0.9, 95% confidence interval (0.5, 1.6)] or DVT [Relative Risk 0.8, 95% confidence interval (0.5, 1.3)]. Efficacy across subgroups, including age, gender, renal function, body mass index (BMI), extent of index PE, location of DVT thrombus, and prior parenteral heparin use was generally consistent (see Figure 4).
Apixaban efficacy in initial treatment of VTE was consistent between patients who were treated for a PE [Relative Risk 0.9, 95% confidence interval (0.5, 1.6)] or DVT [Relative Risk 0.8, 95% confidence interval (0.5, 1.3)]. Efficacy across subgroups, including age, gender, renal function, body mass index (BMI), extent of index PE, location of DVT thrombus, and prior parenteral heparin use was generally consistent (see Figure 4).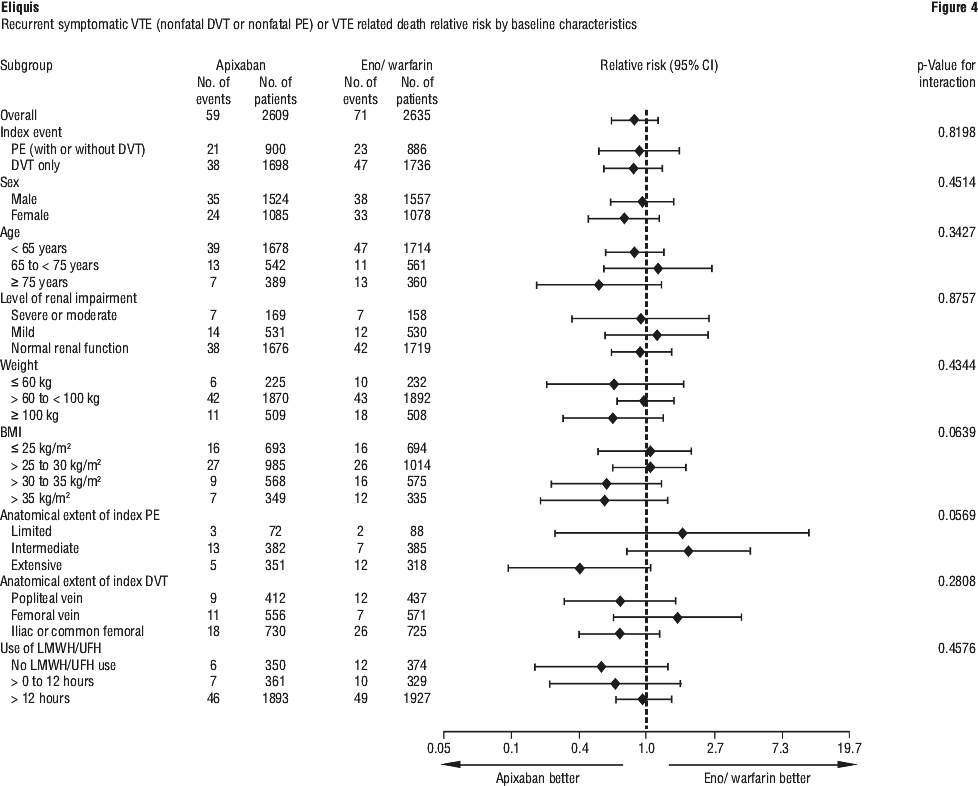 The primary safety endpoint was major bleeding. In the study, apixaban was statistically superior to enoxaparin/ warfarin in the primary safety endpoint [Relative Risk 0.31, 95% confidence interval (0.17, 0.55), p-Value < 0.0001] (see Table 19).
The primary safety endpoint was major bleeding. In the study, apixaban was statistically superior to enoxaparin/ warfarin in the primary safety endpoint [Relative Risk 0.31, 95% confidence interval (0.17, 0.55), p-Value < 0.0001] (see Table 19).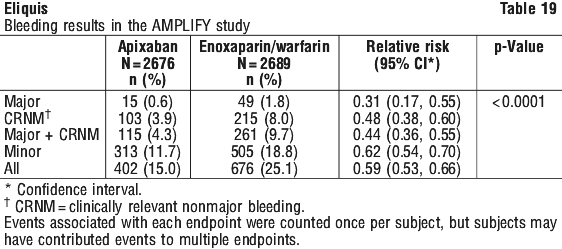 The adjudicated major bleeding and CRNM bleeding at any anatomical site was generally lower in the apixaban group compared to the enoxaparin/ warfarin group. Adjudicated ISTH major gastrointestinal bleeding occurred in 6 (0.2%) apixaban treated patients and 17 (0.6%) enoxaparin/ warfarin treated patients. Adjudicated intracranial bleeding occurred in 3 (0.1%) apixaban treated patients and 6 (0.2%) enoxaparin/ warfarin treated patients.
The adjudicated major bleeding and CRNM bleeding at any anatomical site was generally lower in the apixaban group compared to the enoxaparin/ warfarin group. Adjudicated ISTH major gastrointestinal bleeding occurred in 6 (0.2%) apixaban treated patients and 17 (0.6%) enoxaparin/ warfarin treated patients. Adjudicated intracranial bleeding occurred in 3 (0.1%) apixaban treated patients and 6 (0.2%) enoxaparin/ warfarin treated patients.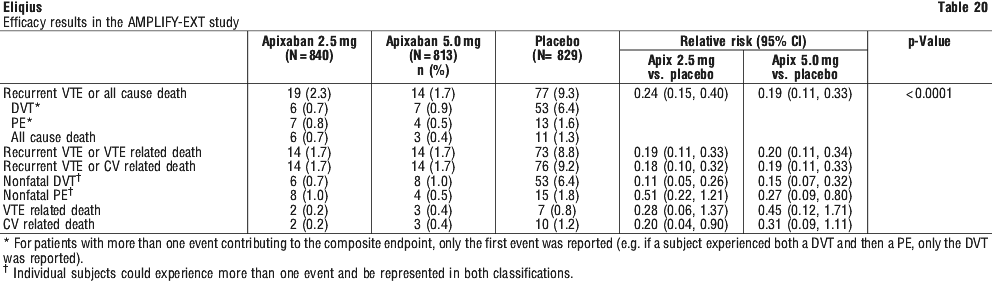 Figure 5 is a plot of the time from randomisation to the occurrence of the first primary efficacy endpoint event in the three treatment groups in the AMPLIFY-EXT study.
Figure 5 is a plot of the time from randomisation to the occurrence of the first primary efficacy endpoint event in the three treatment groups in the AMPLIFY-EXT study.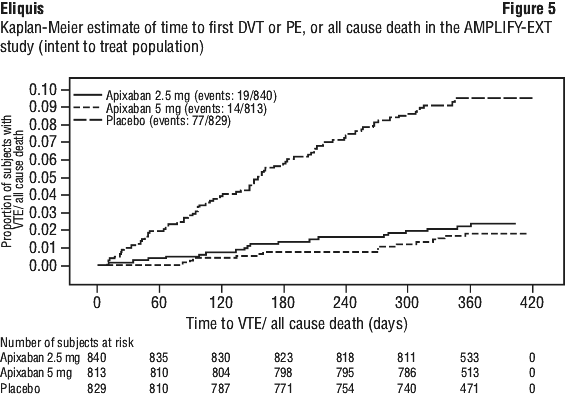 Apixaban efficacy for prevention of a recurrence of a VTE was maintained across subgroups, including age, gender, BMI, and renal function.
Apixaban efficacy for prevention of a recurrence of a VTE was maintained across subgroups, including age, gender, BMI, and renal function.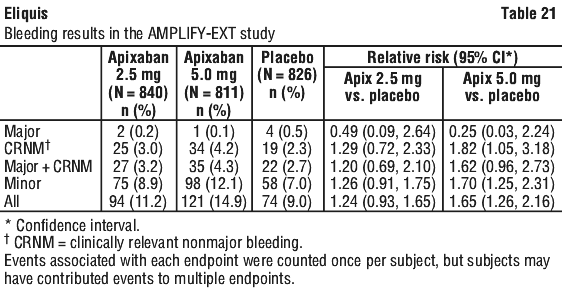 Figure 6 is a plot of the time from randomisation to the occurrence of the first major or clinically relevant nonmajor bleeding event in the three treatment groups in the AMPLIFY-EXT study.
Figure 6 is a plot of the time from randomisation to the occurrence of the first major or clinically relevant nonmajor bleeding event in the three treatment groups in the AMPLIFY-EXT study.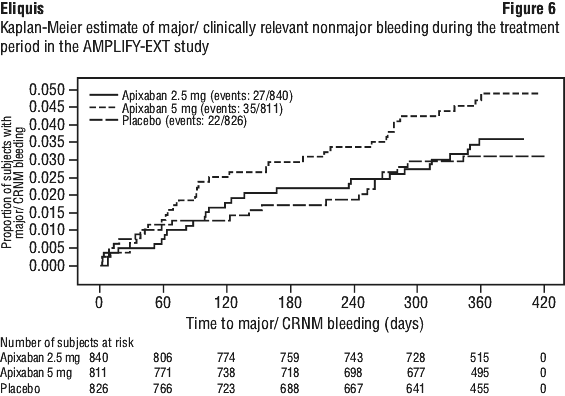 ISTH major gastrointestinal bleeding occurred in 1 (0.1%) apixaban treated patient at the 5 mg twice daily dose, no patients at the 2.5 mg twice daily dose, and 1 (0.1%) placebo treated patient. Adjudicated intracranial bleeding occurred in none of the apixaban treated patients and 1 (0.1%) placebo treated patient.
ISTH major gastrointestinal bleeding occurred in 1 (0.1%) apixaban treated patient at the 5 mg twice daily dose, no patients at the 2.5 mg twice daily dose, and 1 (0.1%) placebo treated patient. Adjudicated intracranial bleeding occurred in none of the apixaban treated patients and 1 (0.1%) placebo treated patient. There was no impact of impaired renal function on peak concentration of apixaban. There was an increase in apixaban exposure correlated to decrease in renal function, as assessed via measured creatinine clearance. In individuals with mild (creatinine clearance 51-80 mL/min), moderate (creatinine clearance 30-50 mL/min) and severe (creatinine clearance 15-29 mL/min) renal impairment, apixaban plasma concentrations (AUC) were increased 16, 29, and 44% respectively, compared to individuals with normal creatinine clearance. Renal impairment had no evident effect on the relationship between apixaban plasma concentration and anti-FXa activity.
There was no impact of impaired renal function on peak concentration of apixaban. There was an increase in apixaban exposure correlated to decrease in renal function, as assessed via measured creatinine clearance. In individuals with mild (creatinine clearance 51-80 mL/min), moderate (creatinine clearance 30-50 mL/min) and severe (creatinine clearance 15-29 mL/min) renal impairment, apixaban plasma concentrations (AUC) were increased 16, 29, and 44% respectively, compared to individuals with normal creatinine clearance. Renal impairment had no evident effect on the relationship between apixaban plasma concentration and anti-FXa activity.