1 Name of Medicine
Elranatamab.
2 Qualitative and Quantitative Composition
Elrexfio 40 mg/mL solution for injection.
Each single-dose vial contains 44 mg of elranatamab in 1.1 mL (40 mg/mL).
Each single-dose vial contains 76 mg of elranatamab in 1.9 mL (40 mg/mL).
Elranatamab is an IgG2 kappa bispecific antibody derived from two monoclonal antibodies (mAbs), an anti-B cell maturation antigen (BCMA) mAb and an anti-Cluster of differentiation 3 (CD3) mAb. Elranatamab is produced using two recombinant Chinese hamster ovary (CHO) cell lines.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection.
The solution is clear to slightly opalescent, colourless to pale brownish liquid solution with a pH of 5.8 and osmolarity of approximately 301 mOsm/L (40 mg/mL solution for injection).
4.1 Therapeutic Indications
Elrexfio has provisional approval in Australia and is indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least 3 prior therapies, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody and have demonstrated disease progression on the last therapy.
The decision to approve this indication has been made on the basis of the overall response rate in a single arm study. Continued approval of this indication depends on verification and description of benefit in confirmatory trials.
4.2 Dose and Method of Administration
Treatment with Elrexfio should be initiated and supervised by physicians experienced in the treatment of multiple myeloma.
Elrexfio should be administered by a healthcare provider with adequately trained medical personnel and appropriate medical equipment to manage severe reactions, including cytokine release syndrome (CRS) and Immune effector cell-associated neurotoxicity syndrome (ICANS) (see Section 4.4 Special Warnings and Precautions for Use).
Dosage.
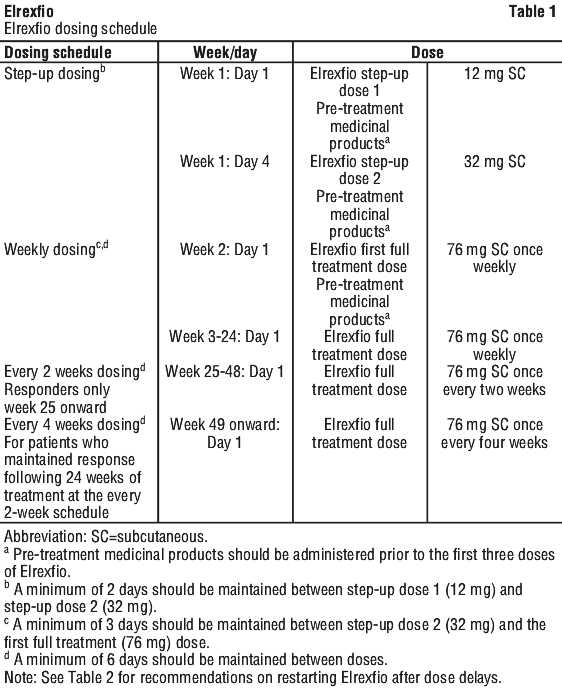
The recommended dosing schedule for Elrexfio is provided in Table 1. The recommended doses of Elrexfio subcutaneous injection are step-up doses of 12 mg on Day 1 and 32 mg on Day 4 followed by a full treatment dose of 76 mg weekly, from Week 2 to Week 24.
For patients who have received at least 24 weeks of treatment with Elrexfio and have maintained a response, the dosing interval should transition to an every two-week schedule. For patients who have received at least 24 weeks of treatment at the every two-week schedule and maintained a response, the dose interval should transition to an every four-week schedule.
Treatment with Elrexfio should be continued until disease progression or unacceptable toxicity.
Method of administration.
Elrexfio is for subcutaneous injection only.
For instructions on handling of the medicinal product before administration, see Section 4.2 Dose and Method of Administration, Instructions for use and handling.
Pre-treatment medicinal products should be administered prior to the first three doses of Elrexfio in the dosing schedule, which includes step-up dose 1 (12 mg), step-up dose 2 (32 mg), and the first full treatment dose (76 mg) as described in Table 1. See Section 4.2 Dose and Method of Administration, Recommended pre-treatment medicinal products.
Hospitalisations and monitoring during initiation.
Due to the risk of severe reactions, hospitalisation is recommended for 48 hours after administration of the first step-up dose, and for 24 hours after administration of the second step-up dose.
For subsequent re-initiations, supervision by a healthcare professional or hospitalisation is recommended.
Elrexfio should be administered subcutaneously according to the step-up dosing schedule in Table 1 to reduce the incidence and severity of CRS and ICANS.
Due to the risk of CRS and ICANS, patients should be monitored for signs and symptoms for 48 hours after administration of each of the 2 step-up doses within the Elrexfio dosing schedule (see Table 3 and 4) (see Section 4.2 Dose and Method of Administration, Hospitalisations and monitoring during initiation; Section 4.4 Special Warnings and Precautions for Use).
Recommended pre-treatment medicinal products.
The following pre-treatment medicinal products should be administered approximately 1 hour prior to the first three doses of Elrexfio in the dosing schedule, which includes step-up dose 1, step-up dose 2, and the first full treatment dose as described in Table 1 to reduce the risk of CRS (see Section 4.4 Special Warnings and Precautions for Use):
paracetamol 500 mg orally (or equivalent);
dexamethasone 20 mg orally or intravenously (or equivalent);
diphenhydramine 25 mg orally (or equivalent).
Missed doses.
If a dose of Elrexfio is missed, the dose should be administered as soon as possible, and the dosing schedule should be adjusted as needed to maintain the dosing interval (see Table 1).
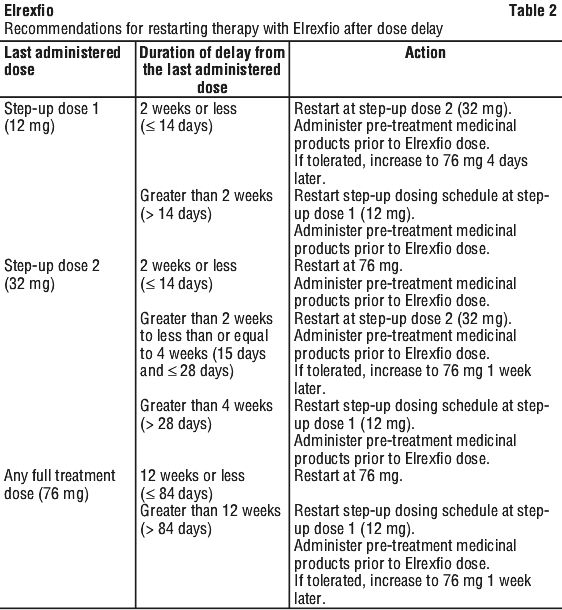
Restarting Elrexfio after dose delay.
If a dose of Elrexfio is delayed, therapy should be restarted based on the recommendations listed in Table 2, and Elrexfio should be resumed according to the dosing schedule (see Table 1).
Pre-treatment medicinal products should be administered as indicated in Table 2.
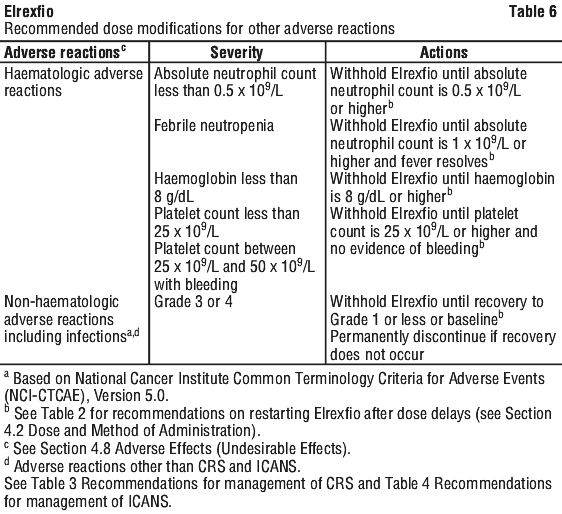
Dosage modifications for Elrexfio.
Dose reductions of Elrexfio are not recommended.
Dose delays may be required to manage toxicities related to Elrexfio (see Section 4.4 Special Warnings and Precautions for Use). Recommendations on restarting Elrexfio after a dose delay are provided in Table 2.
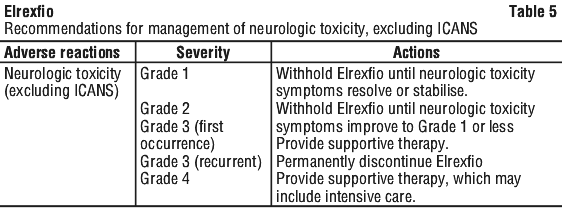
See Tables 3 and 4 for recommended actions for adverse reactions of CRS and ICANS, respectively. See Table 5 for recommended actions for other adverse reactions following administration of Elrexfio.
Cytokine release syndrome (CRS).
Elrexfio can cause CRS, including life-threatening or fatal reactions. Therapy should be initiated according to the step-up dosing schedule to reduce risk of CRS (see Table 1 and Table 2).
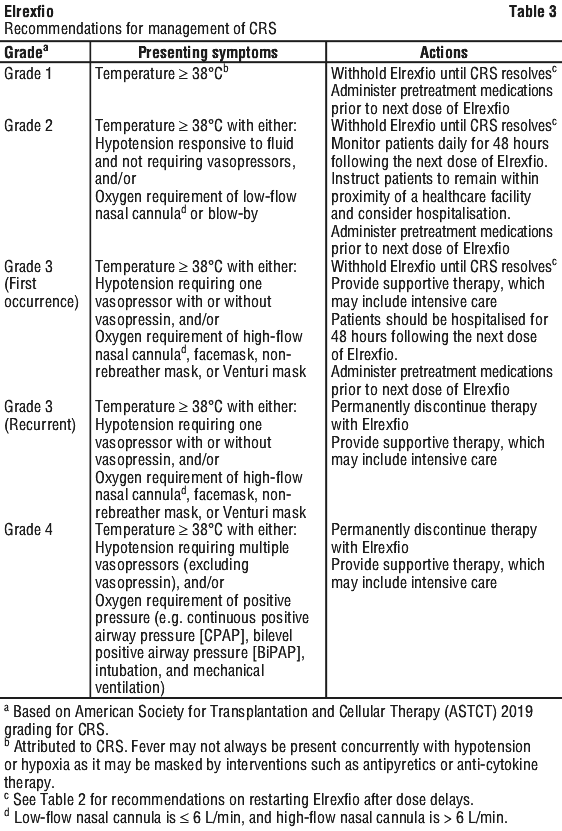
CRS should be identified based on clinical presentation (see Section 4.4 Special Warnings and Precautions for Use). Patients should be evaluated and treated for other causes of fever, hypoxia, and hypotension.
Supportive therapy for CRS (including but not limited to anti-pyretic agents, intravenous fluid support, vasopressors, IL-6 or IL-6 receptor inhibitors, supplemental oxygen, etc.) should be administered as appropriate. Laboratory testing to monitor for disseminated intravascular coagulation (DIC), haematology parameters, as well as pulmonary, cardiac, renal, and hepatic function should be considered.
Management recommendations for CRS are summarised in Table 3.
Neurological toxicities, including immune effector cell-associated neurotoxicity syndrome (ICANS).
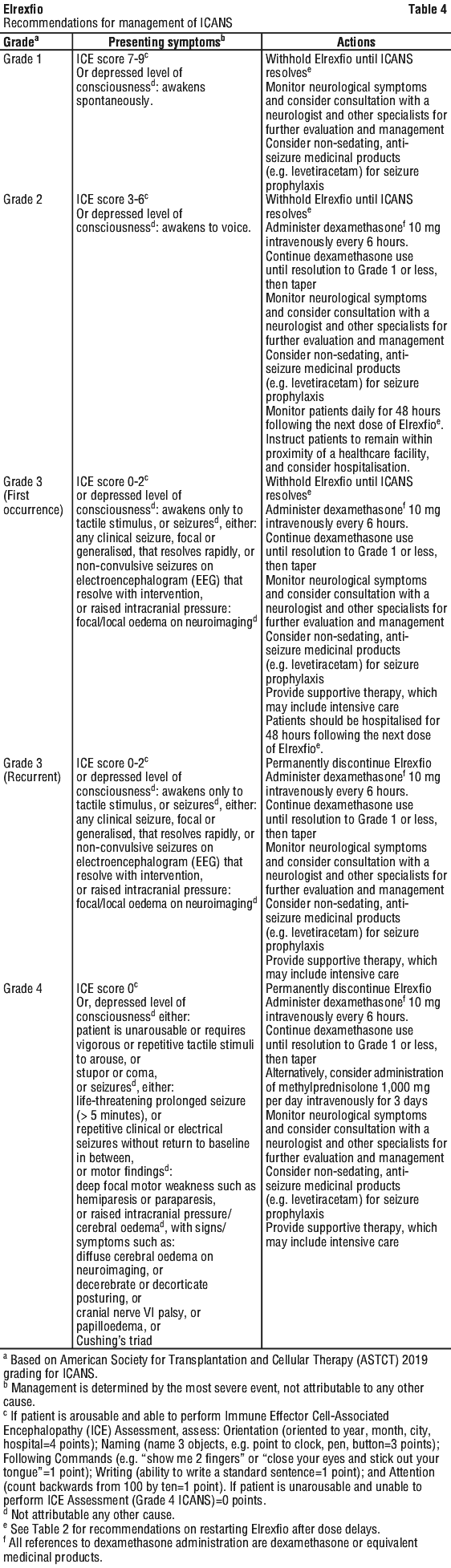
Elrexfio can cause serious or life-threatening neurologic toxicity, including ICANS (see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects)).
At the first sign of neurologic toxicity, including ICANS, withhold Elrexfio and consider immediate neurology evaluation. Other causes of neurological symptoms should be ruled out. Patients should be treated based on severity.
Management recommendations for ICANS and neurological toxicities are summarised in Table 4.
Special populations.
Elderly (65 years of age and older).
No dose adjustment is necessary (see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties).
Renal impairment.
No dose adjustment is recommended in patients with mild to moderate renal impairment. Elrexfio has not been studied in patients with severe renal impairment (eGFR 15 to 29 mL/min) (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
No dose adjustments are required for mild hepatic impairment. The effects of moderate to severe hepatic impairment (total bilirubin > 1.5 times ULN and any AST) on the pharmacokinetics of elranatamab have not been studied (see Section 5.2 Pharmacokinetic Properties).
Paediatric population.
There is no relevant use of Elrexfio in the paediatric population (below 18 years of age) for the treatment of multiple myeloma.
Instructions for use and handling.
Elrexfio 76 mg/1.9 mL (40 mg/mL) vial and 44 mg/1.1 mL (40 mg/mL) vial are supplied as ready-to-use solution. Elrexfio vials do not need dilution prior to administration.
Elrexfio is a clear to slightly opalescent, and colourless to pale brown liquid solution. Elrexfio should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The solution should not be administered if it is discoloured or contains particulate matter.
Aseptic technique should be used to prepare and administer Elrexfio.
Preparation instructions.
Elrexfio is for single use in one patient only. Discard any residue.
Elrexfio should be prepared following the instructions (see Table 7) depending on the required dose.
It is suggested to use a 44 mg/1.1 mL single-dose vial for step-up dose 1 or step-up dose 2.
 Once punctured, the vial and dosing syringe should be used immediately. If the prepared dosing syringe is not used immediately, store syringe at between 2°C to 8°C for a maximum of 24 hours, or at below 30°C for a maximum of 6 hours.
Once punctured, the vial and dosing syringe should be used immediately. If the prepared dosing syringe is not used immediately, store syringe at between 2°C to 8°C for a maximum of 24 hours, or at below 30°C for a maximum of 6 hours.
Administration instructions.
Elrexfio should be administered by a healthcare provider.
The required dose of Elrexfio should be injected into the subcutaneous tissue of the abdomen (preferred injection site). Alternatively, Elrexfio may be injected into the subcutaneous tissue at other sites (e.g. thigh).4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Traceability.
In order to improve the traceability of biological medicinal products, the name and the batch number of the administered medicinal product should be clearly recorded.
Cytokine release syndrome (CRS).
Elrexfio can cause CRS, including life-threatening or fatal reactions.
Clinical signs and symptoms of CRS may include, but are not limited to, fever, hypoxia, chills, hypotension, tachycardia, headache and elevated liver enzymes (see Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be counselled to seek medical attention should signs or symptoms of CRS occur. Patients should also be provided with the Patient Card.
Therapy should be initiated according to Elrexfio step-up dosing schedule (see Table 1) to reduce risk of CRS and patients should be monitored following administration of Elrexfio accordingly (see Section 4.2 Dose and Method of Administration, Hospitalisations and monitoring during initiation). Pre-treatment medicinal products should be administered prior to the first three doses of Elrexfio in the dosing schedule to reduce risk of CRS (see Table 1 and Table 2; see Section 4.2 Dose and Method of Administration).
Management of cytokine release syndrome.
At the first sign of CRS, Elrexfio should be withheld and patients should be immediately evaluated for hospitalisation. CRS should be managed according to the recommendations in Table 3 and further management should be considered per local institutional guidelines (see Section 4.2 Dose and Method of Administration). Supportive therapy for CRS (including but not limited to anti-pyretic agents, intravenous fluid support, vasopressors, IL-6 or IL-6 receptor inhibitors, supplemental oxygen, etc.) should be administered as appropriate. Laboratory testing to monitor for disseminated intravascular coagulation (DIC), haematology parameters, as well as pulmonary, cardiac, renal, and hepatic function should be considered.
General management for CRS is summarised in Table 3 (see Section 4.2 Dose and Method of Administration). Patients who experience Grade 2 or higher CRS with the previous dose of Elrexfio should be instructed to remain within proximity of a healthcare facility and hospitalisation should be considered for 48 hours following the next dose.
Neurological toxicities, including ICANS.
Elrexfio can cause serious or life-threatening neurological toxicities, including ICANS. Clinical signs and symptoms associated with neurologic toxicity included headache, encephalopathy, motor dysfunction, sensory neuropathy, and Guillain-Barré syndrome (see Section 4.8 Adverse Effects (Undesirable Effects)).
Therapy should be initiated according to Elrexfio step-up dosing schedule (see Table 1) to reduce risk of ICANS and patients should be monitored following administration of Elrexfio accordingly (see Section 4.2 Dose and Method of Administration, Hospitalisations and monitoring during initiation).
Patients should be monitored for signs and symptoms (e.g. decrease level of consciousness, seizures and/or motor weakness) of neurologic toxicities during treatment. Patients should be counselled to seek medical attention should signs or symptoms of neurological toxicity occur. Patients should also be provided with the Patient Card.
Due to the potential for ICANS, patients should be advised not to drive or operate heavy or potential dangerous machinery during the Elrexfio step-up dosing schedule and for 48 hours after completing each of the 2 step-up doses within the Elrexfio dosing schedule and in the event of new onset of any neurological symptoms (see Section 4.2 Dose and Method of Administration; Section 4.7 Effects on Ability to Drive and Use Machines).
Management of neurological toxicities, including ICANS.
At the first sign of neurological toxicity, including ICANS, Elrexfio should be withheld and patients should be immediately evaluated and treated based on severity. Supportive therapy, which may include intensive care, for severe or life-threatening neurological toxicities, should be provided.
General management for neurological toxicity (e.g. ICANS) is summarised in Table 4 (see Section 4.2 Dose and Method of Administration). Patients who experience Grade 2 or higher ICANS with the previous dose of Elrexfio should be instructed to remain within proximity of a healthcare facility and hospitalisation should be considered for 48 hours following the next dose.
Infections.
Elrexfio can cause severe, life-threatening, or fatal infections (see Section 4.8 Adverse Effects (Undesirable Effects)). New or reactivated viral infections occurred during and/or following therapy with Elrexfio, including cytomegalovirus (CMV) infection/reactivation. Progressive multifocal leukoencephalopathy (PML), which can be fatal, has been reported during therapy with Elrexfio.
Treatment with Elrexfio should not be initiated in patients with active infections. Patients should be monitored for signs and symptoms of infection prior to and during treatment with Elrexfio and treated appropriately. Elrexfio should be withheld or permanently discontinued based on severity as indicated in Table 6 (see Section 4.2 Dose and Method of Administration). Prophylactic antimicrobials and anti-virals should be administered according to local institutional guidelines. Treatment with subcutaneous or intravenous immunoglobulin (IVIG) should be considered, as appropriate.
Neutropenia.
Elrexfio can cause neutropenia and febrile neutropenia (see Section 4.8 Adverse Effects (Undesirable Effects)).
Complete blood cell counts should be monitored at baseline and periodically during treatment. Supportive therapy should be provided according to local institutional guidelines. Patients with neutropenia should be monitored for signs of infection.
Treatment with Elrexfio should be withheld as indicated in Table 5 (see Section 4.2 Dose and Method of Administration).
Hypogammaglobulinaemia.
Elrexfio can cause Hypogammaglobulinaemia (see Section 4.8 Adverse Effects (Undesirable Effects)).
Immunoglobulin levels should be monitored during treatment with Elrexfio. Subcutaneous or intravenous or immunoglobulin (IVIG) therapy should be considered if IgG levels fall below 400 mg/dL and patients should be treated according to local institutional guidelines, including infection precautions and antimicrobial prophylaxis.
Concomitant use of live viral vaccines.
The safety of immunisation with live viral vaccines during or following treatment with Elrexfio has not been studied. Vaccination with live virus vaccines is not recommended within 4 weeks prior to the first dose of Elrexfio and during treatment with Elrexfio.
Hepatotoxicity.
Elrexfio can cause hepatotoxicity (see Section 4.8 Adverse Effects (Undesirable Effects)). Liver enzyme elevation can occur with or without concurrent CRS.
Monitor liver enzymes and bilirubin at baseline and during treatment as clinically indicated. Elrexfio should be withheld or permanently discontinued based on severity as indicated in Table 6 (see Section 4.2 Dose and Method of Administration).
Use in hepatic impairment.
No formal studies of Elrexfio in patients with hepatic impairment have been conducted. See Section 5.2 Pharmacokinetic Properties.
Use in renal impairment.
No formal studies of Elrexfio in patients with renal impairment have been conducted. See Section 5.2 Pharmacokinetic Properties.
Use in the elderly.
No overall differences in safety or effectiveness were observed between patients ≥ 65 and ≥ 75 years of age compared to younger patients. No dose adjustment required in patients aged ≥ 65 years.
Paediatric use.
The safety and effectiveness of Elrexfio in paediatric patients have not been established.
Effects on laboratory tests.
See Section 4.8 Adverse Effects (Undesirable Effects).4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed with Elrexfio.
Elrexfio causes release of cytokines that may suppress activity of cytochrome P450 (CYP) enzymes, resulting in increased exposure of CYP substrates. Increased exposure of CYP substrates is more likely to occur after the first dose of Elrexfio and up to 14 days after a subsequent step-up dose, as well as during and after CRS.
During this time period, toxicity or medicinal product concentrations (e.g. cyclosporine) should be monitored in patients who are receiving concomitant sensitive CYP substrates with a narrow therapeutic index. The dose of the concomitant medicinal product should be adjusted as needed.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no data on the effect of elranatamab on human fertility and no animal fertility studies have been conducted. In a general toxicity study in cynomolgus monkeys, no changes in the male and female reproductive tract to suggest potential impairment of fertility were observed with elranatamab at subcutaneous doses up to 6 mg/kg/week (yielding exposure 6.5 times greater than in patients at the maximum recommended human dose, based on serum AUC).
Women of childbearing potential/ contraception in females.
Women of childbearing potential should use effective contraception during treatment with Elrexfio and for 4 months after the last dose.
(Category C)
There are no data on the use of Elrexfio in pregnant women. No embryofetal development studies have been conducted with elranatamab in animals.
Based on the mechanism of action, Elrexfio may cause fetal harm when administered to a pregnant woman therefore is not recommended for use during pregnancy. The pregnancy status of women of childbearing potential should be verified prior to initiating treatment with Elrexfio.
Human immunoglobulin (IgG) is known to cross the placenta after the first trimester of pregnancy. Based on the mechanism of action, elranatamab may cause fetal harm, including B-cell lymphocytopenia, when administered to a pregnant woman. Fetal B-cell depletion poses a risk of opportunistic infections in the neonate. Elranatamab-induced cytokine release may also pose a risk for embryofetal loss.
Elrexfio is associated with hypogammaglobulinaemia (see Section 4.4 Special Warnings and Precautions for Use, Hypogammaglobulinaemia), therefore, assessment of immunoglobulin levels in newborns of mothers treated with Elrexfio should be considered.
Postponing vaccination with live or live-attenuated vaccines is recommended for neonates and infants who have been exposed to elranatamab in utero until B-cell levels have recovered.
It is not known whether elranatamab is excreted in human or animal milk, affects breastfed infants or affects milk production. Human IgGs are known to be excreted in breast milk. A risk to the breastfed child cannot be excluded and therefore breast-feeding is not recommended during treatment with Elrexfio and for 4 months after the last dose.4.7 Effects on Ability to Drive and Use Machines
Elrexfio may have major influence on the ability to drive and use machines.
Due to the potential for ICANS, patients receiving Elrexfio are at risk of depressed level of consciousness (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be instructed to refrain from driving or operating heavy or potential dangerous machinery during and for 48 hours after completing each of the 2 step-up doses within the Elrexfio dosing schedule and in the event of new onset of neurological toxicity until resolution of any neurological symptoms (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
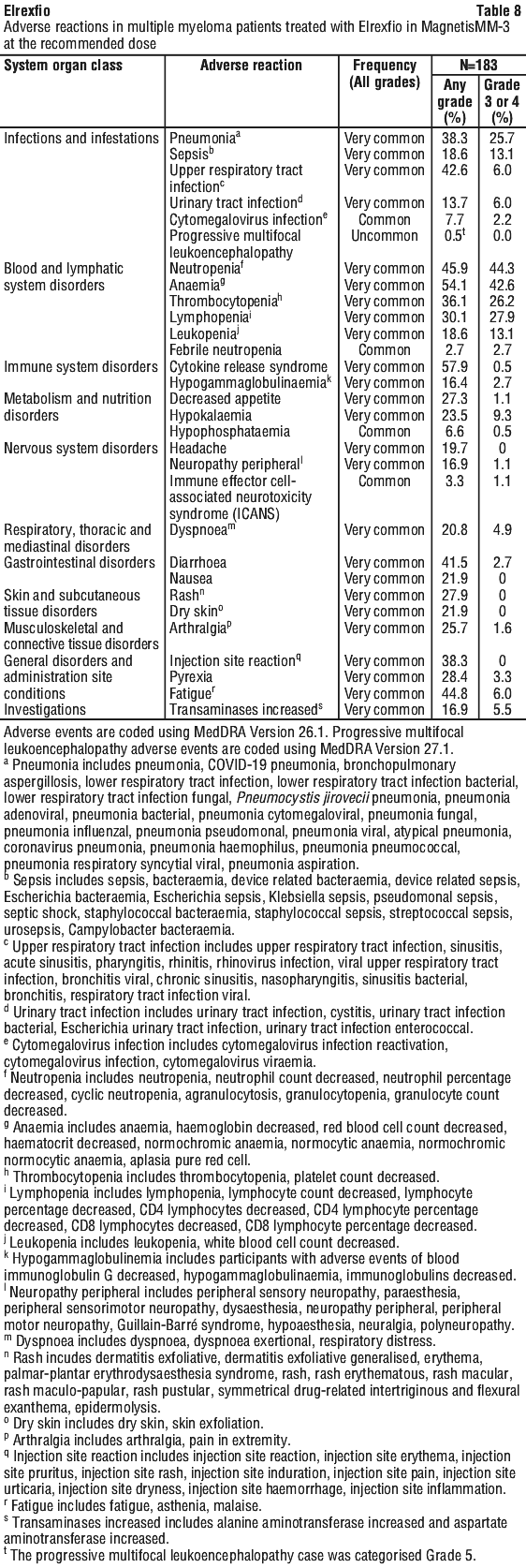
The safety of Elrexfio was evaluated in MagnetisMM-3 (see Section 5.1 Pharmacodynamic Properties, Clinical trials), which included 183 adult patients with multiple myeloma who received the recommended dosing regimen of Elrexfio. The median (range) duration of Elrexfio treatment was 4.1 (0.03 to 31.3) months.
The most frequent adverse reactions of any grade in patients were CRS (57.9%), anaemia (54.1%), neutropenia (45.9%), fatigue (44.8%), upper respiratory tract infection (42.6%), diarrhoea (41.5%), injection site reaction (38.3%), pneumonia (38.3%), thrombocytopenia (36.1%), lymphopenia (30.1%), pyrexia (28.4%), rash (27.9%), decreased appetite (27.3%), arthralgia (25.7%), hypokalaemia (23.5%), nausea (21.9%), dry skin (21.9%) and dyspnoea (20.8%).
Serious adverse reactions included pneumonia (31.7%), sepsis (15.8%), CRS (12.6%), anaemia (5.5%), upper respiratory tract infection (5.5%), urinary tract infection (3.8%), febrile neutropenia (2.7%), diarrhoea (2.7%), dyspnoea (2.2%) and pyrexia (2.2%).
Tabulated list of adverse reactions.
Table 8 summarises adverse reactions reported in patients who received Elrexfio at the recommended dosing regimen (N=183 including 64 patients with prior BCMA-directed antibody drug conjugate [ADC] or chimeric antigen receptor [CAR] T cell therapy [supportive Cohort B]). The safety data of Elrexfio was also evaluated in the all-treated population (N=265) with no additional adverse reactions identified.
Adverse reactions observed during clinical studies are listed by frequency category. Frequency categories are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000) and very rare (< 1/10,000).
Within each frequency grouping, where relevant, adverse reactions are presented in order of decreasing seriousness.
Description of selected adverse reactions.
Cytokine release syndrome (CRS).
CRS occurred in 57.9% of patients who received Elrexfio at the recommended dosing schedule (see Section 4.2 Dose and Method of Administration), with Grade 1 CRS in 43.7% of patients, Grade 2 CRS in 13.7% of patients and Grade 3 CRS in 0.5% of patients. Most patients experienced CRS after the first step-up dose (43.2%) or the second step-up dose (19.1%), with 7.1% of patients having CRS after the first full treatment dose and 1.6% of patients after a subsequent dose. Recurrent CRS occurred in 13.1% of patients. The median time to onset of CRS was 2 (range: 1 to 9) days after the most recent dose, with a median duration of 2 (range: 1 to 19 days) days.
Among patients who developed CRS, associated symptoms included fever (99.0%), hypoxia (11.4%), and hypotension (21.0%). Among patients who developed CRS, 34% received tocilizumab (or siltuximab) and 15.1% received corticosteroids for treatment of CRS.
Immune effector cell-associated neurotoxicity syndrome (ICANS).
ICANS occurred in 3.3% of patients following treatment with Elrexfio at the recommended dosing schedule (see Section 4.2 Dose and Method of Administration), with Grade 1 ICANS in 0.5%, Grade 2 in 1.6% and Grade 3 in 1.1% of patients. The majority of patients had ICANS after the first step-up dose (2.7%), 1 (0.5%) patient had ICANS after the second step-up dose and 1 (0.5%) patient had ICANS after a subsequent dose. Recurrent ICANS occurred in 1.1% of patients. The median time to onset was 3 (range: 1 to 4) days after the most recent dose with a median duration of 2 (range: 1 to 18) days.
The onset of ICANS can be concurrent with CRS, following resolution of CRS, or in the absence of CRS. The most frequent symptoms of ICANS included a depressed level of consciousness and Grade 1 or Grade 2 Immune Effector Cell-Associated Encephalopathy (ICE) scores. Among patients who developed ICANS, 66.7% received corticosteroids, 33.3% received tocilizumab (or siltuximab), 33.3% received levetiracetam and 16.7% received anakinra for treatment of ICANS.
Immunogenicity.
In the MagnetisMM-3 study, of the 168 participants who received recommended step-up and full dosage of Elrexfio for up to 36 months and are evaluable for presence of anti-drug antibodies (ADA) against elranatamab, 9.5% (16/168) of patients tested positive for anti-elranatamab antibodies. Among the 16 patients who tested positive for ADAs, 56% (9/16) tested positive for neutralising antibodies against elranatamab. The effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of Elrexfio products is unknown.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There has been minimal experience with elranatamab overdose in the clinical trial program, and the maximum tolerated dose has not been determined. In the elranatamab clinical trial program, one participant received double the intended dose of elranatamab (accidental overdose), which resulted in hospitalisation for symptoms consistent with cytokine storm. The events resolved following treatment.
In clinical studies, doses up to 76 mg once weekly have been administered.
Treatment.
In the event of an overdose, the patient should be monitored for any signs or symptoms of adverse reactions and appropriate supportive treatment should be instituted immediately.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Elranatamab is a bispecific antibody that binds B-cell maturation antigen (BCMA) expressed on plasma cells, plasmablasts, and multiple myeloma cells and CD3 expressed on T cells. Simultaneous binding of BCMA and CD3 by elranatamab leads to T-cell activation and proliferation and the release of pro-inflammatory cytokines, resulting in the lysis of BCMA-expressing tumour and normal cells.
Pharmacodynamic effects.
Exposure-response relationships.
Serum concentrations of cytokines (IL-2, IL-6, IL-8, IL-10, TNF-α, and IFN-γ) were measured before and after administration of step-up dose 1, step-up dose 2, and the first three full treatment doses of Elrexfio. Time of the maximum cytokine concentration generally occurred during the step-up dosing and concentrations continue to decrease over the course of the first month of treatment.
Clinical trials.
Relapsed or refractory multiple myeloma.
The efficacy of Elrexfio monotherapy was evaluated in patients with relapsed or refractory multiple myeloma in an open-label, non-randomised, multi-centre, Phase 2 study (MagnetisMM-3). The study included patients who were refractory to at least one proteasome inhibitor (PI), one immunomodulatory agent (IMiD), and one anti-CD38 monoclonal antibody. MagnetisMM-3 included 123 patients naïve to prior BCMA-directed therapy (pivotal Cohort A) and 64 patients with prior BCMA directed antibody drug conjugate (ADC) or chimeric antigen receptor (CAR) T cell therapy (supportive Cohort B). Patients had measurable disease by International Myeloma Working Group (IMWG) criteria at enrollment. The study included patients with an ECOG score of ≤ 2, adequate baseline bone marrow (absolute neutrophil count ≥ 1.0 x 109/L, platelet count ≥ 25 x 109/L, haemoglobin level ≥ 8 g/dL), renal (CrCl ≥ 30 mL/min), and hepatic (AST and ALT ≤ 2.5 x ULN, total bilirubin ≤ 2 x ULN) function, and left-ventricular ejection fraction ≥ 40%. Patients with a stem cell transplant within 12 weeks prior to enrollment and active infections were excluded from the study.
Eligible patients received subcutaneous administration of Elrexfio at step-up doses of 12 mg on Day 1 and 32 mg on Day 4 of treatment, followed by the first full treatment dose of Elrexfio (76 mg) on Day 8 of treatment. Thereafter, patients received 76 mg once weekly. After 24 weeks, in patients who achieved an IMWG response category of partial response (PR) or better with responses persisting for at least 2 months, the dosing interval was changed from every week to every 2 weeks. After at least 24 weeks of every 2 weeks dosing, the dosing interval was changed from every 2 weeks to every 4 weeks (see Section 4.2 Dose and Method of Administration).
Among the 187 patients treated with at least 1 dose of elranatamab, 77 (41.2%) switched from every-week dosing to every-2-week dosing and 35 (18.7%) further switched from every-2-week dosing to every-4-week dosing.
Among the 123 patients treated in pivotal Cohort A, the median age was 68 (range: 36 to 89) years with 20% of patients ≥ 75 years of age. 44.7% were female; 58.5% were White, 13.0% were Asian, 8.9% were Hispanic/Latino, and 7.3% were Black. Disease stage (R-ISS) at study entry was 22.8% in Stage I, 55.3% in Stage II, and 15.4% in Stage III. The median time since initial diagnosis of multiple myeloma to enrolment was 72.9 (range: 16 to 228) months. Patients had received a median of 5 prior lines of therapy (range: 2 to 22); with 96.0% who had received ≥ 3 prior lines of therapy. 96.7% were triple-class refractory, and 95.9% refractory to their last line of therapy. 68.3% received prior autologous stem cell transplantation, and 5.7% received prior allogenic stem cell transplantation. High-risk cytogenetics (t(4;14), t(14;16), or del(17p)) were present in 25.2% of patients. 31.7% of patients had extramedullary disease (presence of any plasmacytoma [extramedullary and/or paramedullary] with a soft-tissue component) at baseline by Blinded Independent Central Review (BICR).
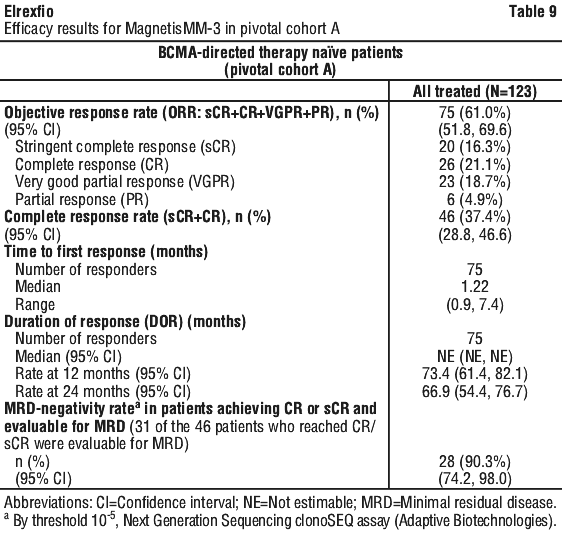
Efficacy results were based on response rate and duration of response (DOR), as assessed by BICR based on the IMWG criteria. Efficacy results from pivotal Cohort A with a data cutoff of 26 March 2024 are shown in Table 9. The median (range) follow-up for responders was 27.9 (3.6, 36.8) months.
 Among the 64 patients treated in supportive Cohort B (BCMA-exposed patients: BCMA-directed ADC and/or CAR T cell therapy), the median age was 67 (range: 41 to 84) years with 18.8% of patients ≥ 75 years of age. 53.1% were female; 68.8% were White, 10.9% were Hispanic/Latino, 3.1% were Black, and 1.6% were Asian. Disease stage (R-ISS) at study entry was 17.2% in Stage I, 56.3% in Stage II and 23.4% in Stage III. The median time since initial diagnosis of multiple myeloma to enrolment was 102.6 (range: 23 to 219) months. Patients had received a median of 7.5 prior lines of therapy (range: 3 to 19); 96.9% were triple-class refractory and 51.6% were penta-drug refractory (refractory to at least 2 PIs, 2 IMiDs, and 1 anti-CD38 antibody); 87.5% were refractory to their last line of therapy. 71.9% and 32.8% received prior ADC and CAR T cell therapy, respectively. 82.8% received prior autologous stem cell transplantation, and 3.1% received prior allogenic stem cell transplantation. High-risk cytogenetics (t(4;14), t(14;16),or del(17p)) were present in 20.3% of patients. 57.8% of patients had extramedullary disease at baseline by BICR.
Among the 64 patients treated in supportive Cohort B (BCMA-exposed patients: BCMA-directed ADC and/or CAR T cell therapy), the median age was 67 (range: 41 to 84) years with 18.8% of patients ≥ 75 years of age. 53.1% were female; 68.8% were White, 10.9% were Hispanic/Latino, 3.1% were Black, and 1.6% were Asian. Disease stage (R-ISS) at study entry was 17.2% in Stage I, 56.3% in Stage II and 23.4% in Stage III. The median time since initial diagnosis of multiple myeloma to enrolment was 102.6 (range: 23 to 219) months. Patients had received a median of 7.5 prior lines of therapy (range: 3 to 19); 96.9% were triple-class refractory and 51.6% were penta-drug refractory (refractory to at least 2 PIs, 2 IMiDs, and 1 anti-CD38 antibody); 87.5% were refractory to their last line of therapy. 71.9% and 32.8% received prior ADC and CAR T cell therapy, respectively. 82.8% received prior autologous stem cell transplantation, and 3.1% received prior allogenic stem cell transplantation. High-risk cytogenetics (t(4;14), t(14;16),or del(17p)) were present in 20.3% of patients. 57.8% of patients had extramedullary disease at baseline by BICR.
Efficacy results in supportive Cohort B for the data cutoff 26 March 2024 include confirmed ORR by BICR of 34.4% (95% CI: 22.9, 47.3); 14.1% of patients achieved CR or better, and 32.8% achieved VGPR or better. Median TTR was 1.92 (range: 0.92, 6.74) months. After a median (range) follow-up of 27.2 (6.41, 29.70) months in responders, median DOR (months) was not reached (95% CI: 12.0, NE). The Kaplan-Meier DOR rate was 69.8% (44.5, 85.2) at 24 months.
5.2 Pharmacokinetic Properties
Pharmacokinetic parameters are presented as geometric mean (coefficient of variation [CV]%) and are based upon subcutaneously administered unless otherwise specified.
The Cmax and AUCtau of elranatamab after the first subcutaneous dose increased in a dose proportional manner over the evaluated dose range via SC administration (~ 6 to 76 mg). The median accumulation ratio after 24 weeks of weekly dosing (steady state) relative to the first subcutaneous dose of elranatamab 76 mg for Cmax and AUCtau was 6.6-fold and 11.2-fold, respectively. The predicted Cmax, Ctrough, and Cavg from the population PK model for the recommended dosage of elranatamab are presented in Table 10.

Absorption.
The predicted mean bioavailability of elranatamab was 56.2% when administered subcutaneously. The median Tmax after elranatamab SC administration across all dose levels ranged from 3 to 7 days for both total and free serum concentrations.
Distribution.
Based on the population pharmacokinetic model, the mean (coefficient of variation [CV]%) central volume of distribution of elranatamab was 4.78 L (69%). The mean peripheral volume of distribution of elranatamab was 2.83 L.
Metabolism.
Elranatamab is expected to be metabolised via normal protein degradation pathways for IgG molecules.
Elimination.
The predicted geometric mean half-life of elranatamab is 22, 64% (CV) days at week 24 following doses of 76 mg weekly. Based on the population pharmacokinetic model, the predicted mean elranatamab clearance was 0.324 L/day, 100% (CV).
Special populations.
No clinically relevant differences in the pharmacokinetics of elranatamab were observed for age (36 to 89 years), sex (167 male, 154 female), race (193 White, 49 Asian, 29 Black), and body weight (37 to 160 kg).
Renal impairment.
No studies of elranatamab in patients with renal impairment have been conducted. Results of population pharmacokinetic analyses indicate that mild renal impairment (60 mL/min/1.73 m2 ≤ estimated glomerular filtration rate (eGFR) < 90 mL/min/1.73 m2) or moderate renal impairment (30 mL/min/1.73 m2 ≤ eGFR < 60 mL/min/1.73 m2) did not significantly influence the pharmacokinetics of elranatamab. Limited data are available from patients with severe (eGFR less than 30 mL/min/1.73 m2) renal impairment.
Hepatic impairment.
No studies of elranatamab in patients with hepatic impairment have been conducted. Results of population pharmacokinetic analyses indicate that mild hepatic impairment (total bilirubin > 1 to 1.5 times upper limit of normal (ULN) and any aspartate aminotransferase (AST), or total bilirubin ≤ ULN and AST > ULN) did not significantly influence the pharmacokinetics of elranatamab. No data are available in patients with moderate (total bilirubin > 1.5 to 3.0 x ULN and any AST) or severe (total bilirubin > 3.0 x ULN and any AST) hepatic impairment.
5.3 Preclinical Safety Data
Genotoxicity.
No genotoxicity studies have been performed with elranatamab. As a large protein molecule, elranatamab is not expected to interact directly with DNA or other chromosomal material.
Carcinogenicity.
No animal studies have been performed to assess the carcinogenic potential of elranatamab.6 Pharmaceutical Particulars
6.1 List of Excipients
Histidine, histidine hydrochloride monohydrate, disodium edetate, polysorbate 80, sucrose, water for injections.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
Unopened vial.
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
Prepared syringe.
The prepared syringe should be administered immediately. If the prepared dosing syringe is not used immediately, store syringe at between 2°C to 8°C for a maximum of 24 hours, or at below 30°C for a maximum of 6 hours.
6.4 Special Precautions for Storage
Store in a refrigerator (2°C to 8°C).
Do not freeze. Do not shake.
Store in the original carton in order to protect from light.
For storage conditions after first opening of the medicinal product, see Section 6.3 Shelf Life.
6.5 Nature and Contents of Container
1.1 mL solution in a single-dose vial (Type 1 glass) with a stopper (butyl rubber) and an aluminium seal with a flip-off cap containing 44 mg of elranatamab.
Pack size of 1 vial.
1.9 mL solution in a single-dose vial (Type 1 glass) with a stopper (butyl rubber) and an aluminium seal with a flip-off cap containing 76 mg of elranatamab.
Pack size of 1 vial.
6.6 Special Precautions for Disposal
The vial and any remaining contents after withdrawal of a single-dose should be discarded. In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
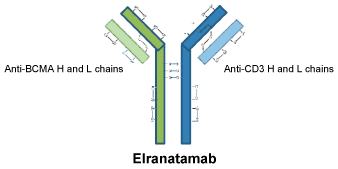 Elranatamab is a bispecific IgG2 kappa antibody derived from two monoclonal antibodies (mAbs), the anti-BCMA mAb and the anti-CD3 mAb. Each of these mAbs contributes one distinct heavy (H) chain and one distinct light (L) chain to the bispecific elranatamab. The resulting 4-chain bispecific antibody is covalently linked via five inter-chain disulfide bonds.
Elranatamab is a bispecific IgG2 kappa antibody derived from two monoclonal antibodies (mAbs), the anti-BCMA mAb and the anti-CD3 mAb. Each of these mAbs contributes one distinct heavy (H) chain and one distinct light (L) chain to the bispecific elranatamab. The resulting 4-chain bispecific antibody is covalently linked via five inter-chain disulfide bonds.
CAS number.
2408850-14-4.7 Medicine Schedule (Poisons Standard)
Schedule 4 (Prescription Only Medicine).
Summary Table of Changes
