1 Name of Medicine
Pegcetacoplan.
2 Qualitative and Quantitative Composition
Each 20 mL vial contains 1,080 mg pegcetacoplan in a pH 5.0, 10 mmol acetate buffer.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection.
Clear, colourless to slightly yellowish aqueous solution, practically free from visible particles, to be administered by subcutaneous infusion.
Empaveli solution for injection does not contain any antimicrobial preservatives. The vial is for single use in one patient on one occasion only. Discard any residue.
4.1 Therapeutic Indications
Empaveli is indicated in the treatment of adult patients with paroxysmal nocturnal haemoglobinuria (PNH).
Empaveli is indicated for the treatment of adults and adolescents aged 12 to 17 years with C3 glomerulopathy (C3G) or primary immune-complex membranoproliferative glomerulonephritis (IC-MPGN).
4.2 Dose and Method of Administration
Recommended vaccination and prophylaxis.
Before receiving treatment with Empaveli. Patients with known history of vaccination.
Ensure that patients have received vaccines against encapsulated bacteria including Streptococcus pneumoniae, Neisseria meningitidis types A, C, W, Y, and B, and Haemophilus influenzae type B (Hib) within 2 years prior to starting Empaveli (see Section 4.4 Special Warnings and Precautions for Use).
Patients without known history of vaccination.
Administer required vaccines at least 2 weeks prior to receiving the first dose of Empaveli (see Section 4.4 Special Warnings and Precautions for Use).
If immediate therapy with Empaveli is indicated, administer required vaccines as soon as possible and provide patients with antibacterial drug prophylaxis until 2 weeks after vaccination (see Section 4.4 Special Warnings and Precautions for Use).
PNH, C3G and primary IC-MPGN are chronic diseases and treatment with Empaveli is recommended to continue for the patient's lifetime, unless the discontinuation of Empaveli is clinically indicated (for monitoring of PNH manifestations, see Section 4.4 Special Warnings and Precautions for Use).
Dosage.
Empaveli can be given by a healthcare professional or administered by the patient or caregiver following proper instructions.
PNH. Adult patients with PNH.
Empaveli is administered twice weekly as a 1,080 mg subcutaneous infusion with a commercially available syringe system infusion pump that can deliver doses up to 20 mL (see Method of administration). The twice weekly dose should be administered on Day 1 and Day 4 of each treatment week. After proper training in subcutaneous infusion, a patient may self-administer, or the patient's caregiver may administer Empaveli, if a healthcare provider determines that it is appropriate.
Switching to Empaveli from a C5 inhibitor (eculizumab rmc, ravulizumab rch).
For the first 4 weeks, Empaveli is administered as twice weekly subcutaneous doses of 1,080 mg in addition to the patient's current dose of C5 inhibitor treatment to minimise the risk of haemolysis with abrupt treatment discontinuation. After 4 weeks, the patient should discontinue treatment with the C5 inhibitor before continuing on monotherapy with Empaveli.
Dose adjustment for PNH treatment with Empaveli.
The dosing regimen may be changed to 1,080 mg every third day if a subject has a lactate dehydrogenase (LDH) level greater than 2 x upper limit of normal (ULN).
In the event of a dose increase, monitor LDH twice weekly for at least 4 weeks.
C3G and primary IC-MPGN. Empaveli is administered twice weekly as a subcutaneous infusion with a commercially available syringe system infusion pump that can deliver doses up to 20 mL. The twice weekly dose should be administered on Day 1 and Day 4 of each treatment week (see Method of administration).
Adult patients with C3G or primary IC-MPGN.
Empaveli is administered twice weekly as a 1,080 mg subcutaneous infusion.
Adolescent patients with C3G or primary IC-MPGN.
For adolescent patients, the dosing regimen is based on the patient's body weight and consists of the following (see Table 1):
 Missed dose of Empaveli. If a dose of Empaveli is missed, and the next scheduled dose has not been administered, the missed dose should be administered as soon as possible, and the regular schedule resumed even if this results in an interval of less than 3 days between the replacement dose and the subsequent dose.
Missed dose of Empaveli. If a dose of Empaveli is missed, and the next scheduled dose has not been administered, the missed dose should be administered as soon as possible, and the regular schedule resumed even if this results in an interval of less than 3 days between the replacement dose and the subsequent dose.
Two doses should not be administered on the same day; however, it is acceptable to administer doses on 2 consecutive days.
Method of administration.
Empaveli should only be administered via subcutaneous administration using a syringe system infusion pump.
When therapy with Empaveli is initiated, the patient will be instructed by a qualified healthcare provider in infusion techniques, the use of a syringe system infusion pump, the keeping of a treatment record, the recognition of possible adverse reactions, and measures to be taken in case these occur.
Infuse Empaveli in the abdomen, thighs, hips, or upper arms. Infusion sites should be at least 7.5 cm apart from each other. Rotate infusion sites between administration. Do not infuse into areas where the skin is tender, bruised, red, or hard. Avoid infusing into tattoos, scars, or stretch marks.
The typical infusion time is approximately 30 minutes (if using two sites) or approximately 60 minutes (if using one site). The infusion should be started promptly after drawing Empaveli into the syringe. Complete the administration within 2 hours after preparing the syringe.
Renal impairment.
Renal impairment had no effect on the pharmacokinetics of pegcetacoplan; therefore, dose adjustment of Empaveli in patients with renal impairment is not necessary (see Section 5.2 Pharmacokinetic Properties). There are no data available for the use of pegcetacoplan in patients with end-stage renal disease requiring dialysis.
Hepatic impairment.
The safety and efficacy of Empaveli have not been studied in patients with hepatic impairment; however, population pharmacokinetic data suggest that no dose adjustment is required in patients with hepatic impairment (see Section 5.2 Pharmacokinetic Properties).4.3 Contraindications
Empaveli is contraindicated in patients with:
Hypersensitivity to pegcetacoplan or to any of the excipients;
Unresolved infection caused by encapsulated bacteria including Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae (see Section 4.4 Special Warnings and Precautions for Use).
4.4 Special Warnings and Precautions for Use
Serious infections caused by encapsulated bacteria.
The use of Empaveli may predispose individuals to serious infections caused by encapsulated bacteria including Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae. To reduce the risk of infection, all patients must be vaccinated against these bacteria according to current local guidelines at least 2 weeks prior to receiving Empaveli, unless the risk of delaying therapy with Empaveli outweighs the risk of developing an infection. Patients who initiate treatment with Empaveli less than 2 weeks after vaccination must receive treatment with appropriate prophylactic antibiotics until 2 weeks after vaccination.
Vaccination may not be sufficient to prevent serious infection. Consider official guidance on the appropriate use of antibacterial agents. Monitor all patients for early signs of serious infection, evaluate immediately if infection is suspected, and treat with appropriate antibiotics if necessary. Inform patients of these signs and symptoms and that they should seek medical care immediately.
Hypersensitivity.
Hypersensitivity reactions have been reported. If a severe hypersensitivity reaction (including anaphylaxis) occurs, discontinue infusion with Empaveli immediately and institute appropriate treatment.
Monitoring PNH manifestations after discontinuation of Empaveli.
If patients with PNH discontinue treatment with Empaveli, they should be closely monitored for signs and symptoms of serious intravascular haemolysis. Serious intravascular haemolysis is identified by elevated LDH levels along with sudden decrease in PNH clone size or haemoglobin (Hb), or reappearance of symptoms such as fatigue, haemoglobinuria, abdominal pain, shortness of breath (dyspnoea), major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction. If discontinuation of Empaveli is necessary, an alternate therapy should be considered because PNH is life-threatening if untreated. If serious haemolysis occurs after discontinuation, consider the following procedures/treatments: blood transfusion (packed RBCs), exchange transfusion, anticoagulation and corticosteroids. In addition, slow weaning should be considered, and patients should be closely monitored for at least 8 weeks to detect haemolysis and other reactions, as alternative complement inhibitors may not prevent haemolysis as efficiently.
PNH laboratory monitoring.
Patients with PNH receiving Empaveli should be monitored regularly for signs and symptoms of haemolysis, including measuring LDH levels, and may require dose adjustment within the recommended dosing schedule (see Section 4.2 Dose and Method of Administration).
Excipients with known effect.
This medicinal product contains sorbitol. The additive effect of concomitantly administered products containing sorbitol (or fructose) and dietary intake of sorbitol (or fructose) should be taken into consideration for patients known or suspected to have the rare genetic disorder of hereditary fructose intolerance (HFI).
Use in the elderly.
Although there were no apparent age-related differences observed in clinical studies, the number of patients aged 65 years and over was not sufficient to determine whether they respond differently from younger subjects.
Paediatric use.
The safety and efficacy of pegcetacoplan in children with PNH from birth to less than 18 years have not been established. No data are available.
This medicinal product should not be used in children < 12 years of age, as non-clinical safety data are not available for this age group.
The safety and efficacy of pegcetacoplan in children with C3G or primary IC-MPGN from birth to less than 12 years have not been established. No data are available.
Effects on laboratory tests.
There may be interference between silica reagents in coagulation panels and Empaveli that results in artificially prolonged activated partial thromboplastin time (aPTT); therefore, the use of silica reagents in coagulation panels should be avoided.4.5 Interactions with Other Medicines and Other Forms of Interactions
No clinical interaction studies have been performed. Nonclinical studies showed that pegcetacoplan has a low potential for pharmacokinetic drug interactions, as it did not induce or inhibit cytochrome P450 isozyme activities or serve as a substrate and/or inhibitor for human drug transporters.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Effects of pegcetacoplan upon fertility have not been studied in animals. There were no microscopic abnormalities in male or female reproductive organs in toxicity studies in rabbits and monkeys dosed daily subcutaneously for up to 9 months at exposure levels up to 6 times the clinical area under the curve (AUC).
(Category B3)
There are insufficient data on Empaveli use in pregnant women to inform a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
It is recommended that women of childbearing potential use effective contraception methods to prevent pregnancy during treatment with pegcetacoplan and for at least 8 weeks after the last dose of pegcetacoplan. Pregnancy testing is advised for females of reproductive potential prior to treatment with Empaveli.
Use of Empaveli in pregnancy should be carefully considered, with regard to the specific risks of PNH (including maternal and neonatal death and non-live birth) and benefits for each patient. Empaveli should be used during pregnancy only if the potential benefit justifies the potential risk to the mother, fetus, and/or neonate.
Clinical considerations. Disease-associated maternal and/or fetal/neonatal risk.
In pregnancy, PNH is associated with adverse maternal outcomes, including worsening cytopenias, thrombotic events, infections, bleeding, miscarriages and increased maternal mortality, and adverse fetal outcomes, including fetal death and premature delivery.
Animal data. Animal reproduction studies with pegcetacoplan were conducted in rats, rabbits, and cynomolgus monkeys. Pegcetacoplan treatment of pregnant cynomolgus monkeys at a subcutaneous dose of 28 mg/kg/day (2.9 times the human steady-state AUC) from the gestation period through parturition resulted in a statistically significant increase in abortions or stillbirths compared to controls. The relative exposure at the no-adverse-effect level for this effect (7 mg/kg/day) was similar to that anticipated clinically (1.3 times AUC). No maternal toxicity or teratogenic effects were observed in offspring delivered at term. Additionally, no developmental effects were observed in infants up to 6 months postpartum. Minimal systemic exposure to pegcetacoplan (less than 1% of maternal exposure) was detected in fetuses from monkeys treated with 28 mg/kg/day from the period of organogenesis through the second trimester.
It is not known whether pegcetacoplan is secreted in human milk or whether there is potential for absorption and harm to the infant. Animal data and the chemical nature of pegcetacoplan suggest that the risk of clinically relevant exposure to the infant is minimal.
Minimal (less than 1%) pegcetacoplan excretion in milk has been demonstrated in monkeys; therefore, the probability of clinically relevant exposure of breastfed infant through breastmilk is considered low.
It is recommended to discontinue breast-feeding during pegcetacoplan treatment.4.7 Effects on Ability to Drive and Use Machines
Empaveli has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
PNH.
Clinical trial experience in adult patients with PNH. Study in complement inhibitor-experienced adult patients with PNH (Study APL2-302).
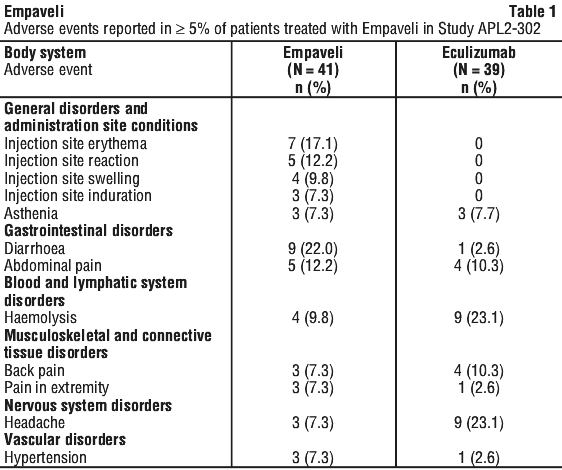
The data described in Table 2 reflect the exposure in 80 adult patients with PNH who received Empaveli (n=41) or eculizumab (n=39) at the recommended dosing regimens for 16 weeks in Study APL2-302.
Serious adverse events were reported in 7 (17%) patients with PNH receiving Empaveli. The most common serious adverse reaction in patients treated with Empaveli was infections (5%).
 Clinically relevant adverse reactions in less than 5% of patients include: intestinal ischemia; biliary sepsis; hypersensitivity pneumonitis.
Clinically relevant adverse reactions in less than 5% of patients include: intestinal ischemia; biliary sepsis; hypersensitivity pneumonitis.
The data described in Table 3 reflect the exposure in the 80 adult patients in Study APL2-302 who received Empaveli at the recommended dosing regimens for up to 48 weeks.

Study in complement inhibitor-naïve adult patients with PNH (Study APL2-308).
The data described below reflect the exposure in adult patients with PNH who received Empaveli (n=46) or the control arm (supportive care excluding complement inhibitors) (n=18) in Study APL2-308. One patient (2%) who received Empaveli died due to septic shock. Serious adverse events were reported in 6 (13%) patients with PNH receiving Empaveli. The most common adverse events (≥ 10%) in patients treated with Empaveli were injection site reactions, infections, viral infection, pain in extremity, hypokalaemia, arthralgia, dizziness, abdominal pain, rash, and headache.
Table 4 describes the adverse events that occurred in ≥ 5% of patients treated with Empaveli in Study APL2-308.
Description of selected adverse reactions.
Injection site reactions.
Injections site reactions (e.g. erythema, swelling, induration, pruritus, and pain) have been reported during Study APL2-302 and APL2-308. These reactions were not severe and did not lead to discontinuation of treatment.
Diarrhoea.
Cases of diarrhoea have been reported during Study APL2-302 and APL2-308, none of them were severe or led to discontinuation of treatment.
Haemolysis.
Haemolysis and haemolytic anaemia have been reported during Study APL2-302 and APL2-308. In Study APL2-302, these events occurred less frequently in the Empaveli group than in the eculizumab group during the randomised controlled period (RCP) (Week 16). There were 3 cases of haemolysis during Study APL2-308 in patients treated with pegcetacoplan. None of these cases were reported as serious or led to discontinuation of pegcetacoplan. The dose of pegcetacoplan was increased in all 3 patients.
C3G or primary IC-MPGN.
Clinical trial experience in patients with C3G or primary IC-MPGN. Study in adolescent and adult patients with C3G or primary IC-MPGN (Study APL2-C3G-310).
The data described below reflects the exposure in (n=63) adolescent and adult patients with C3G or primary IC-MPGN who received Empaveli at the recommended dosing regimens for 26 weeks. Serious adverse events were reported in 6 (10%) patients with C3G or primary IC-MPGN receiving Empaveli. One death was reported due to COVID-19 pneumonia/respiratory failure in an Empaveli-treated patient and was considered as not related to treatment. The most common adverse events (≥ 10%) with Empaveli (adults and adolescents) were infusion site reactions, pyrexia, nasopharyngitis, influenza, cough, and nausea. No Empaveli patient with post-transplant recurrent C3G or primary IC-MPGN experienced kidney transplant rejection or allograft loss.
Table 5 describes the adverse events that occurred in ≥ 5% of patients treated with Empaveli and greater than placebo in Study APL2-C3G-310.
Immunogenicity.
Two different assays for the detection of anti-pegcetacoplan peptide anti-drug antibody (ADA) were used in PNH and C3G or primary IC-MPGN clinical studies, respectively. The assay used for C3G or primary IC-MPGN was more sensitive.
PNH.
Anti-drug antibody incidence (treatment-emergent ADA or boosted ADA from pre-existing level) was low, and when present, had no noticeable impact on the pharmacokinetics/pharmacodynamics (PK/PD), efficacy, or safety profile of pegcetacoplan. Throughout studies APL2-302 and APL2-308, 3 out of 126 patients who were exposed to pegcetacoplan had confirmed positive anti-pegcetacoplan peptide antibodies. All 3 patients also tested positive for neutralising antibody (NAb). NAb response had no apparent impact on PK or clinical efficacy. Eighteen out of 126 patients developed anti-polyethylene glycol (PEG) antibodies; nine were treatment-emergent and nine were treatment-boosted.
C3G and primary IC-MPGN.
ADA incidence (treatment-emergent ADA or boosted ADA from pre-existing level) in Study APL2-C3G-310 was 23.6% for anti-PEG and 16.3% for anti-pegcetacoplan peptide. Based on population PK and PD analysis, ADAs had no clinically meaningful impact on efficacy or PK/PD in a pooled analysis population. Five patients also tested positive for NAb. NAb response had no apparent impact on PK or clinical efficacy. Twenty-nine out of 123 patients developed anti-PEG antibodies; 14 were treatment-emergent and 15 were treatment-boosted. In patients with posttransplant recurrent disease in Study APL2-C3G-204, no patient developed a positive ADA response (treatment-emergent ADA or boosted ADA from pre-existing level) to pegcetacoplan peptide or PEG. During the 26-week placebo-controlled period in Study APL2-C3G-310, there was no detectable impact of ADAs on the safety of pegcetacoplan treatment.
Post marketing experience.
In post marketing experience, the following additional adverse reactions have been reported:
Immune system disorders: anaphylactic reaction, anaphylactic shock.
Skin and subcutaneous tissue disorders: urticaria.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
No case of overdose has been reported during clinical studies.
For information on the management of overdose, contact the Poison Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Selective immunosuppressants, ATC code: LO4AJ03.
Mechanism of action.
Pegcetacoplan binds to complement protein C3 and its activation fragment C3b with high affinity, thereby regulating the cleavage of C3 and the generation of downstream effectors of complement activation. In PNH, extravascular haemolysis (EVH) is facilitated by C3b opsonisation while intravascular haemolysis (IVH) is mediated by the downstream membrane attack complex (MAC). Pegcetacoplan exerts broad regulation of the complement cascade by acting proximal to both C3b and MAC formation, thereby controlling the mechanisms that lead to EVH and IVH. These functions of pegcetacoplan underlie the observed sustained reduction in complement mediated haemolytic activity in patients with PNH.
In C3G and primary IC-MPGN, there is excessive deposition of C3 breakdown products in the glomeruli of the kidney leading to renal parenchymal damage and impairment of kidney function. Pegcetacoplan targets upstream effectors of complement activation (C3 and C3b), thereby inhibiting activation initiated by all (alternative, classical and lectin) complement pathways. By inhibiting C3, pegcetacoplan directly addresses the inappropriate C3 activation and modifies the underlying disease by reducing the excessive deposition of C3 breakdown products in the glomeruli of the kidney. By targeting C3b, pegcetacoplan also inhibits the activity of the alternative pathway (AP) C3 convertase through an additional mechanism of action in the complement cascade. This further prevents deposition of C3 breakdown products in the glomeruli.
Pharmacodynamic effects.
PNH.
In Study APL2-302, the mean serum C3 concentration increased from 0.94 g/L at baseline to 3.83 g/L at Week 16 in the pegcetacoplan group and sustained through Week 48. In Study APL2-308, the mean serum C3 concentration increased from 0.95 g/L at baseline to 3.56 g/L at Week 26.
In Study APL2-302, the percentage of PNH Type II + III RBCs increased from 66.80% at baseline to 93.85% at Week 16 and sustained through Week 48. In Study APL2-308, the mean percentage of PNH Type II + III RBCs increased from 42.4% at baseline to 90.0% at Week 26.
In Study APL2-302, the mean percentage of PNH Type II + III RBCs with C3 deposition was decreased from 17.73% at baseline to 0.20% at Week 16 and sustained through Week 48. In Study APL2-308, the mean percentage of PNH Type II + III RBCs with C3 deposition decreased from 2.85% at baseline to 0.09% at Week 26.
Pegcetacoplan treatment of patients with PNH resulted in the improvement of haemoglobin (Hb) level and reduction of absolute reticulocyte count (ARC) and LDH (see Clinical trials).
C3G and primary IC-MPGN.
In Study APL2-C3G-310, mean serum C3 concentration increased from 0.62 g/L at baseline to 3.71 g/L at Week 26 in the pegcetacoplan group and remained stable (0.57 g/L at baseline; 0.58 g/L at Week 26) in the placebo group.
Mean serum sC5b-9 concentration decreased from 902.5 nanogram/mL at baseline to 290.2 nanogram/mL at Week 26 in the pegcetacoplan group and remained stable (768.3 nanogram/mL at baseline; 759.9 nanogram/mL at Week 26) in the placebo group.
At Week 26, the proportion of patients with an at least two-orders-of-magnitude reduction from baseline in C3c staining intensity on renal biopsy was 74.3% in the pegcetacoplan group, with 71.4% achieving a staining score of zero as compared to 11.8% of patients with a decrease by two orders of magnitude and 8.8% reaching a staining score of zero in the placebo group.
In Study APL2-C3G-204, in patients with post-transplant recurrent disease, mean serum C3 concentration increased from 0.70 g/L at baseline to 2.80 g/L at Week 52, and mean serum sC5b9 concentration decreased from 525.4 nanogram/mL at baseline to 151.0 nanogram/mL at Week 52.
Clinical trials.
PNH. The efficacy and safety of Empaveli in patients with PNH was assessed in two open-label, randomised, controlled, Phase 3 studies (Study APL2-302 (NCT03500549) and Study APL2-308 (NCT04085601)). All patients who completed the studies were eligible to enroll in a separate long-term extension study.
In both studies, patients were vaccinated against Streptococcus pneumoniae, Neisseria meningitidis types A, C, W, Y and B, and Haemophilus influenzae type B (Hib), either within 2 years prior to Day 1 or within 2 weeks after starting treatment with Empaveli. Patients vaccinated after Day 1 received prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination. In addition, prophylactic antibiotic therapy was administered at the discretion of the investigator in accordance with local treatment guidelines for patients with PNH who are receiving treatment with a complement inhibitor.
The dose of Empaveli was 1,080 mg twice weekly. If required, the dose of Empaveli could be adjusted to 1,080 mg every 3 days. Empaveli was administered as a subcutaneous infusion; the infusion time was approximately 20 to 40 minutes.
Study in complement inhibitor-experienced adult PNH patients (APL2-302).
This study enrolled patients with PNH who had been treated with a stable dose of eculizumab for at least the previous 3 months and with Hb levels < 10.5 g/dL. The dose of Empaveli was 1,080 mg twice weekly. Eligible patients entered a 4-week run-in period during which they received Empaveli 1,080 mg subcutaneously twice weekly in addition to their current dose of eculizumab. Patients were then randomised in a 1:1 ratio to receive either 1,080 mg of Empaveli twice weekly or their current dose of eculizumab through the duration of the 16-week randomised controlled period (RCP). If required, the dose of Empaveli could be adjusted to 1,080 mg every 3 days. Randomisation was stratified based on the number of packed red blood cell (PRBC) transfusions within the 12 months prior to Day 28 (< 4; ≥ 4) and platelet count at screening (< 100,000/mm3; ≥ 100,000/mm3). Following completion of the RCP, all patients entered a 32-week open-label period and received monotherapy with Empaveli. All patients who completed the 48-week period were eligible to enroll in a separate long-term extension study.
The primary efficacy endpoint was change from baseline to Week 16 (during RCP) in haemoglobin level. Baseline was defined as the average of measurements recorded prior to taking the first dose of Empaveli. Key secondary efficacy endpoints were transfusion avoidance, defined as the proportion of patients who did not require a transfusion during the RCP, and change from baseline to Week 16 in ARC, LDH level, and the functional assessment of chronic illness therapy (FACIT)-fatigue scale score.
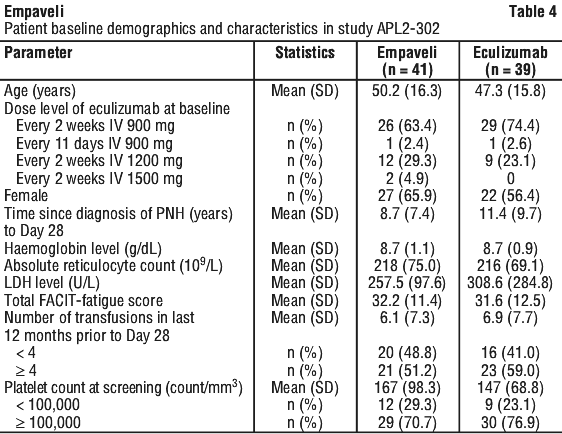
A total of 80 patients were randomised to receive treatment, 41 to Empaveli and 39 to eculizumab. Demographics and baseline disease characteristics were generally well balanced between treatment groups (see Table 6). A total of 38 patients in the group treated with Empaveli and 39 patients in the eculizumab group completed the 16-week RCP and continued into the 32-week open-label period (OLP). Per protocol 15 patients had their dose adjusted to 1,080 mg every three days. Twelve patients were evaluated for benefit and 8 of the 12 patients demonstrated benefit from the dose adjustment.
 Empaveli was superior to eculizumab for the primary endpoint of the haemoglobin change from baseline (P < 0.0001). The adjusted mean change from baseline in Hb level was 2.4 g/dL in the group treated with Empaveli versus -1.5 g/dL in the eculizumab group, demonstrating an adjusted mean increase of 3.8 g/dL with Empaveli compared to eculizumab at Week 16 (Figure 1). Treatment differences between the Empaveli and eculizumab groups were evident as early as Week 2 and persisted throughout the 16-week RCP.
Empaveli was superior to eculizumab for the primary endpoint of the haemoglobin change from baseline (P < 0.0001). The adjusted mean change from baseline in Hb level was 2.4 g/dL in the group treated with Empaveli versus -1.5 g/dL in the eculizumab group, demonstrating an adjusted mean increase of 3.8 g/dL with Empaveli compared to eculizumab at Week 16 (Figure 1). Treatment differences between the Empaveli and eculizumab groups were evident as early as Week 2 and persisted throughout the 16-week RCP.
 The adjusted means, treatment difference, confidence intervals, and statistical analyses performed for the key secondary endpoints are shown in Figure 2.
The adjusted means, treatment difference, confidence intervals, and statistical analyses performed for the key secondary endpoints are shown in Figure 2.
 In patients treated with Empaveli, primary and key secondary efficacy analyses showed no notable differences based on sex, race, or age.
In patients treated with Empaveli, primary and key secondary efficacy analyses showed no notable differences based on sex, race, or age.
Normalisation of ARC was achieved in 78% of patients in the group treated with Empaveli and in 3% in the eculizumab group. LDH normalisation was achieved in 71% of patients in the group treated with Empaveli and in 15% in the eculizumab group.
A total of 77 patients entered the 32-week OLP, during which all patients received Empaveli resulting in a total exposure of up to 48 weeks. The results at Week 48 were generally consistent with those at Week 16 and support sustained efficacy.
Study in complement inhibitor-naïve adult PNH patients (APL2-308).
Study APL2-308 was a randomised, open-label, standard of care (SOC)-controlled study that enrolled patients with PNH who had not been treated with any complement inhibitor within 3 months prior to enrolment and with Hb levels less than the lower limit of normal (LLN). Eligible patients were randomised in a 2:1 ratio to receive Empaveli or SOC excluding complement inhibitors, hereafter referred to as SOC through the duration of the 26-week treatment period.
Randomisation was stratified based on the number of packed red blood cell (PRBC) transfusions within the 12 months prior to Day 28 (< 4; ≥ 4). At any point during the study, a patient assigned to the SOC treatment group who had Hb levels ≥ 2 g/dL below baseline or presented with a PNH associated thromboembolic event was per protocol able to transition to Empaveli for the remainder of the study.
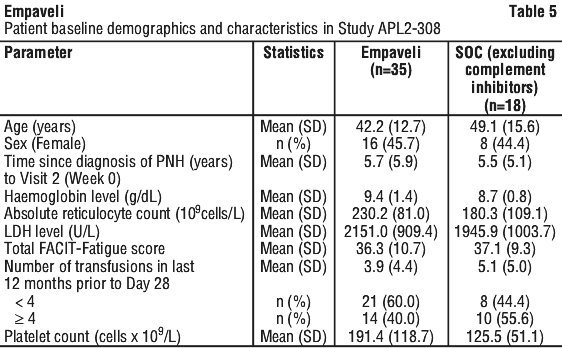
A total of 53 patients were randomised, 35 to Empaveli and 18 patients to SOC. Demographics and baseline disease characteristics were generally well balanced between treatment groups (see Table 7). Eleven of 18 patients randomised to SOC transitioned to Empaveli because their Hb levels decreased by ≥ 2 g/dL below baseline. Per protocol, 3 patients had their dose adjustment to 1,080 mg every three days.
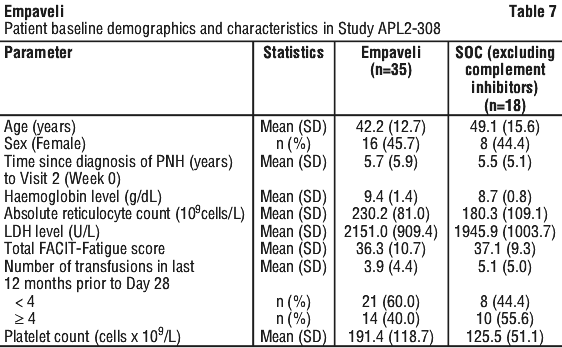 The primary and secondary efficacy endpoints were assessed at Week 26. The two co-primary efficacy endpoints were Hb stabilisation, defined as avoidance of a > 1 g/dL decrease in Hb concentration from baseline in the absence of transfusion, and change in LDH concentration from baseline.
The primary and secondary efficacy endpoints were assessed at Week 26. The two co-primary efficacy endpoints were Hb stabilisation, defined as avoidance of a > 1 g/dL decrease in Hb concentration from baseline in the absence of transfusion, and change in LDH concentration from baseline.
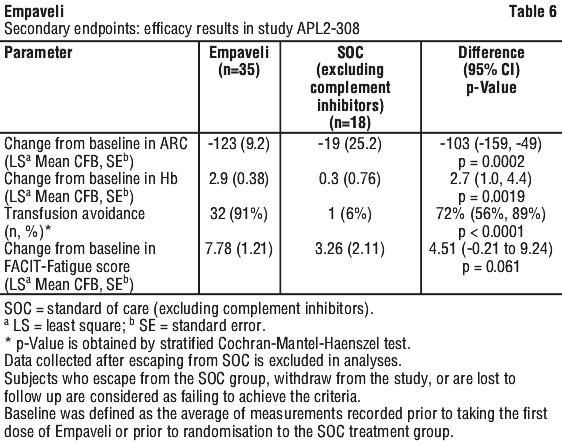
Empaveli was superior to SOC for the first co-primary endpoint of Hb stabilisation through Week 26 (p < 0.0001). In the group treated with Empaveli, 30 out of 35 patients (85.7%) achieved Hb stabilisation versus 0 patients in the SOC group. The adjusted difference between Empaveli and SOC was 73.1% (95% CI, 57.2% to 89.0%).
Empaveli was also superior to SOC for the second co-primary endpoint of change from baseline in LDH concentration at Week 26 (p < 0.0001). The least-square (LS) mean (SE) changes from baseline in LDH were -1870 U/L in the group treated with Empaveli versus -400 U/L in the SOC group. The difference between Empaveli and SOC was -1470 (95% CI, -2113 to -827). Treatment differences between the Empaveli and SOC groups were evident at Week 2 and were maintained through Week 26 (see Figure 3). LDH concentrations in the SOC group remained elevated.
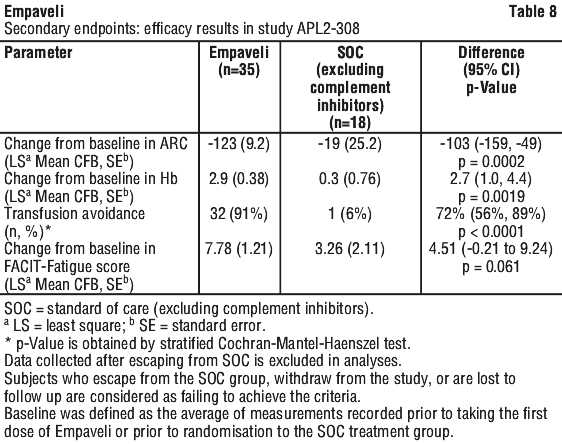
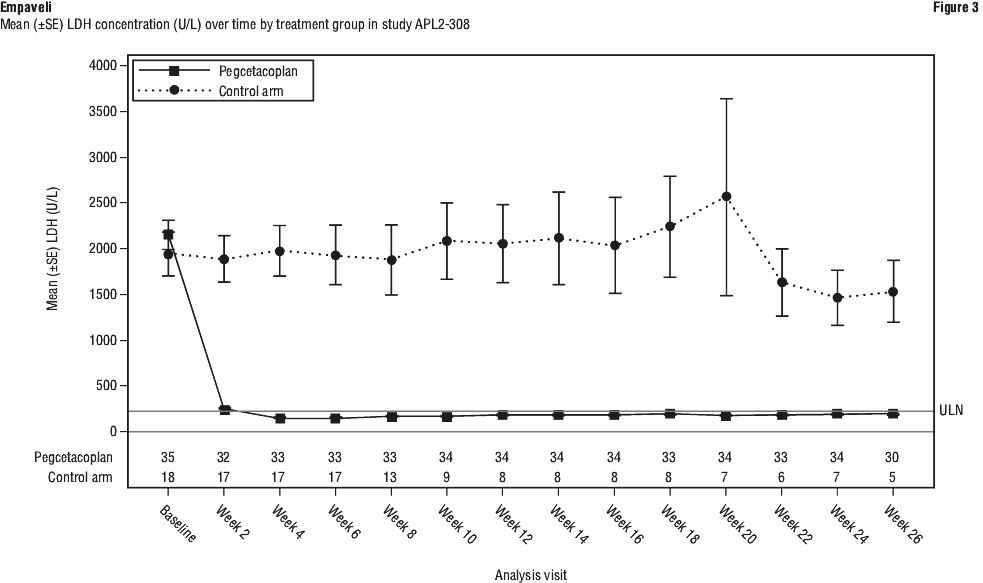 For the secondary efficacy endpoints of change from baseline in ARC, change from baseline in Hb and transfusion avoidance, superiority was demonstrated for Empaveli versus SOC.
For the secondary efficacy endpoints of change from baseline in ARC, change from baseline in Hb and transfusion avoidance, superiority was demonstrated for Empaveli versus SOC.
In the group treated with Empaveli, the mean change from baseline in ARC was -123 x 109/L versus -19 x 109/L in the SOC, demonstrating an adjusted mean decrease of 104 x 109/L compared to SOC.
In the group treated with Empaveli, the mean change from baseline in Hb was 2.94 g/dL versus 0.27 g/dL in the SOC, demonstrating an adjusted mean difference of 2.67 g/dL compared to SOC.
Transfusion avoidance was achieved in 91% of patients in the group treated with Empaveli, as compared to 6% in the SOC group.
Superiority was not demonstrated for the change from baseline in FACIT-fatigue score, however the adjusted mean change from baseline in FACIT-fatigue score was 7.8 points in the group treated with Empaveli versus 3.3 points in the SOC group, demonstrating an adjusted mean increase of 4.5 points compared to SOC.
The adjusted means, treatment differences, confidence intervals and statistical analyses performed for the selected secondary endpoints are shown in Table 8.
 C3G and primary IC-MPGN. The efficacy and safety of Empaveli in patients with C3G or primary IC-MPGN was assessed in the randomised, placebo-controlled, double-blinded Phase 3 Study APL2C3G-310, including adults and adolescents with native kidney or post-transplant recurrent C3G or primary IC-MPGN, and in the open-label, randomised-controlled Phase 2 Study APL2-C3G-204, including adults with post-transplant recurrent C3G or primary IC-MPGN. There is limited data on the safety and efficacy of Empaveli in patients with recurrent IC-MPGN following kidney transplant.
C3G and primary IC-MPGN. The efficacy and safety of Empaveli in patients with C3G or primary IC-MPGN was assessed in the randomised, placebo-controlled, double-blinded Phase 3 Study APL2C3G-310, including adults and adolescents with native kidney or post-transplant recurrent C3G or primary IC-MPGN, and in the open-label, randomised-controlled Phase 2 Study APL2-C3G-204, including adults with post-transplant recurrent C3G or primary IC-MPGN. There is limited data on the safety and efficacy of Empaveli in patients with recurrent IC-MPGN following kidney transplant.
Patients were vaccinated against Streptococcus pneumoniae, Neisseria meningitidis types A, C, W, Y, and B, and Haemophilus influenzae type B (Hib) prior to starting treatment with Empaveli.
The dose of Empaveli was 1,080 mg twice weekly for adults or adolescents with body weights ≥ 50 kg, or weight-based for adolescents with body weights < 50 kg.
Study in adult and adolescent patients with C3G or primary IC-MPGN (APL2-C3G-310).
Study APL2-C3G-310 was a randomised, double-blinded study with a placebo-controlled period of 26-weeks, followed by a 26-week open-label period. This study enrolled adult and adolescent patients aged 12 years and older and weighing at least 30 kg, with biopsy-proven native kidney or post-transplant recurrent C3G or primary IC-MPGN, eGFR ≥ 30 mL/min/1.73m2, proteinuria ≥ 1 g/day, and urine protein-to-creatinine ratio (uPCR) ≥ 1 g/g, and no more than 50% global glomerulosclerosis or interstitial fibrosis on baseline biopsy. For at least 12 weeks before randomisation and throughout the 26-week placebo-controlled period, patients were required to be on stable and optimised doses of angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and/or sodium-glucose cotransporter-2 (SGLT-2) inhibitors. Immunosuppressant medication doses (e.g. systemic corticosteroids no higher than 20 mg prednisone-equivalent daily, mycophenolate mofetil, tacrolimus) had to be stable for at least 12 weeks before randomisation and throughout the 26-week placebo-controlled period.
Eligible patients were randomised in a 1:1 ratio to receive Empaveli or placebo subcutaneously twice weekly during the 26-week RCP. Two stratification factors were applied to the randomisation; patients with post-transplant recurrence versus native kidney disease patients, and patients with baseline renal biopsies (either collected during screening or within 28 weeks prior to randomisation) versus patients without baseline renal biopsies. Patients who completed the RCP, entered the 26-week OLP, in which all participants were treated with Empaveli twice weekly. Only data from the 26-week RCP are described below.
A total of 124 patients were randomised, 63 to Empaveli and 61 to placebo. Demographics and baseline disease characteristics were generally balanced between the two groups (see Table 9).
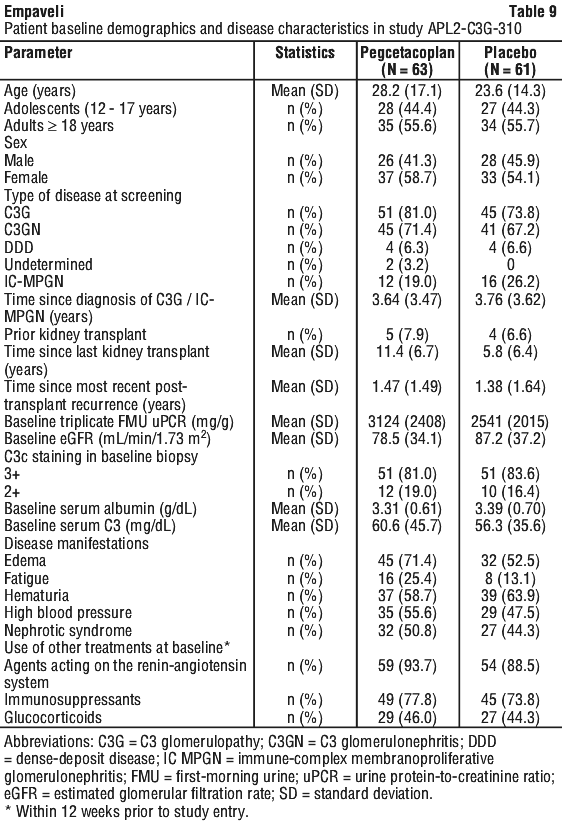 The primary and key secondary efficacy endpoints were assessed at Week 26. The primary efficacy endpoint was the log-transformed ratio of first-morning urine (FMU) protein-to-creatinine ratio (uPCR) at Week 26 compared with baseline.
The primary and key secondary efficacy endpoints were assessed at Week 26. The primary efficacy endpoint was the log-transformed ratio of first-morning urine (FMU) protein-to-creatinine ratio (uPCR) at Week 26 compared with baseline.
Treatment with Empaveli demonstrated a statistically significant percent reduction in uPCR at Week 26 compared to placebo.
Similar reductions were observed in subgroups by age, disease type, transplant status and concomitant use of immunosuppressants/glucocorticoids (Table 10). Subgroup analyses should be interpreted with caution due to a limited number of participants including a limited number with post-transplant disease recurrent primary IC-MPGN.
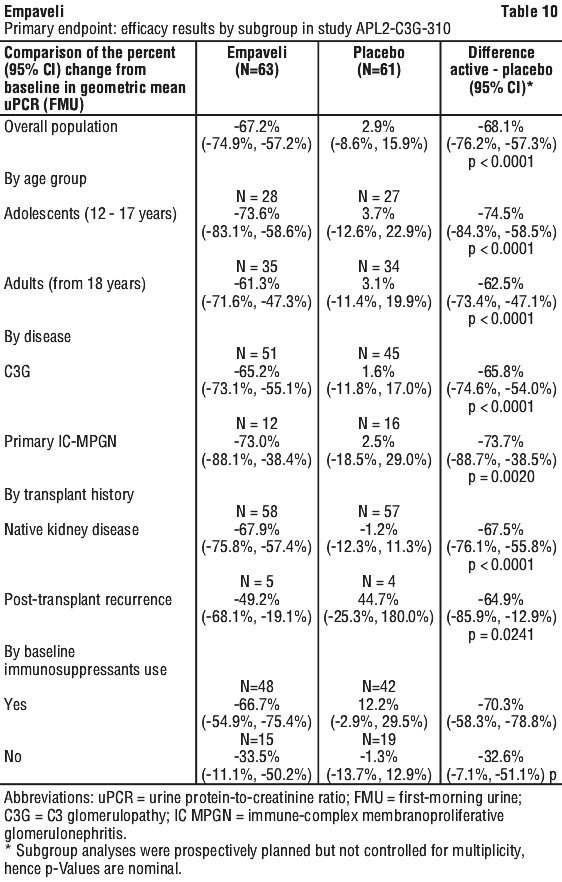 The reduction in proteinuria was apparent as early as Week 4 (Figure 4).
The reduction in proteinuria was apparent as early as Week 4 (Figure 4).
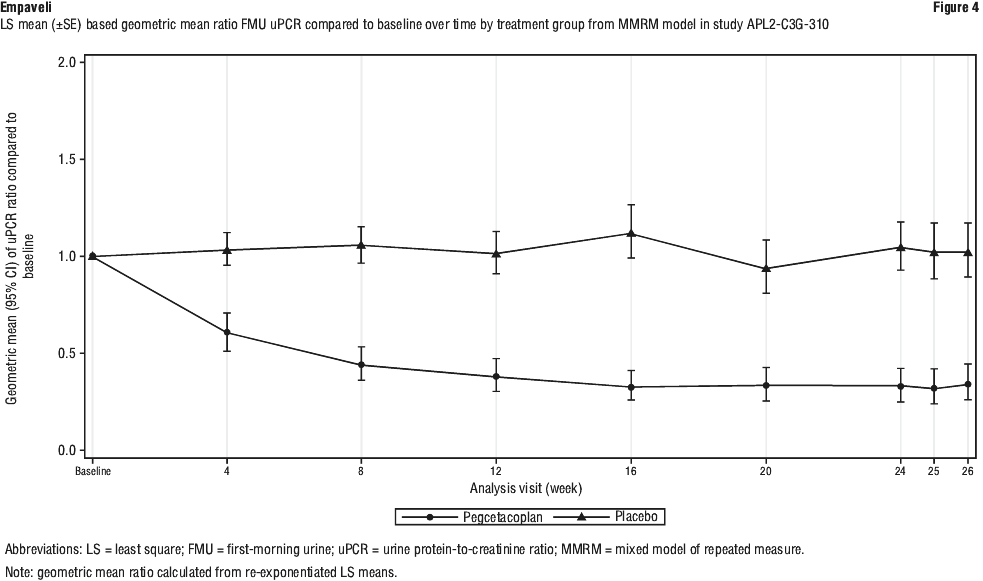 Treatment with Empaveli also demonstrated statistically significant improvement in the key secondary endpoints related to proteinuria reduction. Whilst pegcetacoplan was potentially disease-modifying as indicated by the reduction in C3c staining intensity on renal biopsy and potential stabilisation of estimated glomerular filtration rate (eGFR), this has not been proven. Statistical significance of eGFR stabilisation has not been established. Improvements in total activity score of the C3G histologic index identified in the pegcetacoplan group were not statistically significant. Whilst C3c staining intensity on renal biopsy appeared to reduce in the pegcetacoplan group, magnitude of C3c staining that would correlate with improved outcomes is unclear and only a nominal p-Value is available.
Treatment with Empaveli also demonstrated statistically significant improvement in the key secondary endpoints related to proteinuria reduction. Whilst pegcetacoplan was potentially disease-modifying as indicated by the reduction in C3c staining intensity on renal biopsy and potential stabilisation of estimated glomerular filtration rate (eGFR), this has not been proven. Statistical significance of eGFR stabilisation has not been established. Improvements in total activity score of the C3G histologic index identified in the pegcetacoplan group were not statistically significant. Whilst C3c staining intensity on renal biopsy appeared to reduce in the pegcetacoplan group, magnitude of C3c staining that would correlate with improved outcomes is unclear and only a nominal p-Value is available.
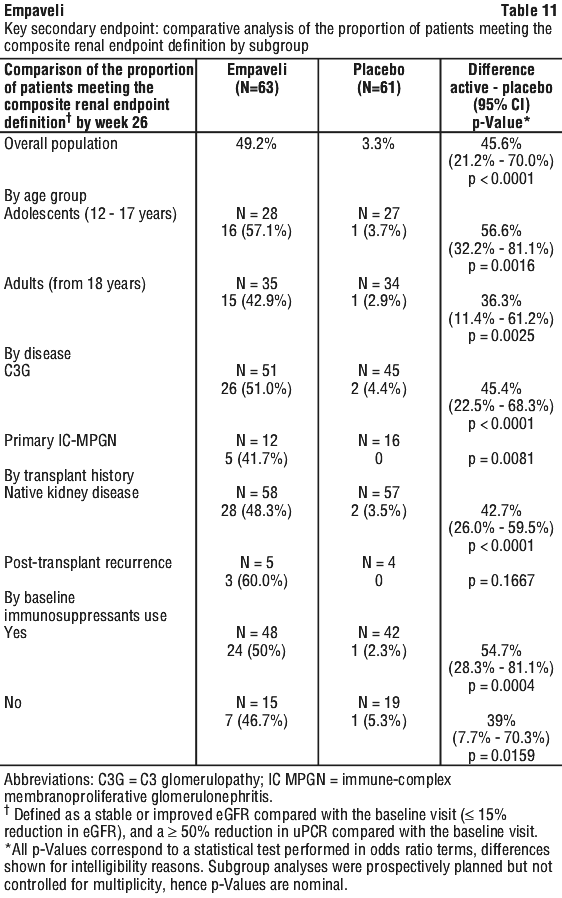
These effects on the key secondary endpoints were consistent in subgroups by age, disease type, transplant status and concomitant use of immunosuppressants/glucocorticoids. Subgroup analyses for the key secondary endpoints at Week 26 are provided in Table 11 to Table 14. Subgroup analyses should be interpreted with caution due to a limited number of participants including a limited number with post-transplant disease recurrent primary IC-MPGN.



Study in patients with post-transplant recurrent C3G or primary IC-MPGN (APL2-C3G-204).
Study APL2-C3G-204 was a Phase 2 open-label, randomised study in adult patients with posttransplant recurrent C3G or primary IC-MPGN.
Eligible patients were randomised in a 3:1 ratio to receive Empaveli in addition to SoC or maintain SoC treatment for 12 weeks and then all patients in the study received Empaveli from Week 13 to Week 52.
The primary efficacy endpoint was the proportion of patients with reduction in C3c staining intensity on renal biopsy after 12 weeks of treatment. Reduction was defined as a decrease by at least two orders of magnitude in visual staining.
Secondary efficacy endpoints included the proportion of patients with reduction in C3c staining intensity on renal biopsy after 52 weeks of treatment, the proportion of patients with stabilisation or improvement in eGFR, and the changes and percentage changes from baseline in eGFR over time.
A total of 13 patients (10 with C3G and 3 with primary IC-MPGN) were randomised, 10 to Empaveli and 3 patients to SoC. Demographics and baseline disease characteristics were generally well balanced between treatment groups. All 13 patients completed the 12-week controlled part, of which 10 completed treatment with Empaveli up to Week 52.
Reduction in C3c staining intensity on renal biopsy at Week 12 was observed in 50% of the patients treated with Empaveli (5 of 10 patients; 95% CI, 18.7%-81.3%; 4 of these patients had a staining score of zero), and 33.3% of the patients in the control group (1 of 3 patients; 95% CI, 0.8-90.6, this patient had a staining score of 1). At Week 52, a reduction in C3c staining intensity on renal biopsy was observed in 53.8% of patients overall (7 of 13 patients; 95% CI, 25.1-80.8). Among those, 6 patients achieved a C3c staining score of zero by Week 52.
In general, changes and percentage changes from baseline in eGFR were small. Mean (SD) eGFR changed from 52.3 (12.11) mL/min/1.73 m2 at baseline to 57.3 (25.12) mL/min/1.73 m2 at Week 52, and median eGFR changed from 50.5 mL/min/1.73 m2 at baseline to 58.5 mL/min/1.73 m2 at Week 52. Most patients (9 of 13 patients [69.2%]) across groups achieved stabilisation or improvement in eGFR by Week 52.
This study evaluated pegcetacoplan in 13 patients and was designed to explore the efficacy of pegcetacoplan in post-transplant recurrent disease evidence for the overall benefit of pegcetacoplan in patients with post-transplant recurrent C3G or primary IC-MPGN is supported by results from this study and from the post-transplant recurrence subgroup of Study APL2-C3G-310.
5.2 Pharmacokinetic Properties
Absorption.
Empaveli is administered subcutaneously and is gradually absorbed into the systemic circulation with a median Tmax between 108 and 144 hours (4.5 to 6.0 days). Steady-state serum concentrations following twice weekly dosing at 1,080 mg in PNH patients were achieved approximately 4 to 6 weeks following the first dose and therapeutic concentrations of pegcetacoplan were maintained through Week 48.
Steady-state serum concentrations following twice weekly dosing at 1,080 mg in C3G or primary IC-MPGN patients were achieved approximately 4 to 8 weeks following the first dose and therapeutic concentrations of pegcetacoplan were maintained through Week 52.
Distribution.
The mean (%CV) of central volume of distribution of pegcetacoplan is approximately 3.98 L (32%) in patients with PNH.
The mean (%CV) of central volume of distribution of pegcetacoplan is approximately 4.31 L (32.1%) in adult patients with C3G or primary IC-MPGN.
Metabolism/excretion.
Based on its PEGylated peptide structure, the metabolism of pegcetacoplan is expected to occur via catabolic pathways and be degraded into small peptides and amino acids.
Following multiple subcutaneous dosing of pegcetacoplan, the estimated mean (CV%) of clearance (CL) is 0.015 L/hour (30%) and median effective half-life of elimination (t1/2) is 8.6 days in patients with PNH.
The estimated mean (CV%) of CL is 0.012 L/hour (43%) in adult patients with C3G or primary IC-MPGN. The median terminal t1/2 is 10.1 days in adult patients with C3G or primary IC-MPGNI.
Results of a study of radiolabeled pegcetacoplan in cynomolgus monkeys suggest the primary route of elimination is via urinary excretion.
Pegcetacoplan showed no inhibition or induction of the CYP enzyme isoforms tested as demonstrated from the results of in vitro studies. Pegcetacoplan was neither a substrate not an inhibitor of the human uptake or efflux transporters.
Linearity/non-linearity.
Exposure of pegcetacoplan increases in a dose-proportional manner from 45 to 1440 mg.
Special populations.
No impact on the pharmacokinetics of pegcetacoplan was identified with age, sex, race, and hepatic function based on the results of population PK analysis in patient with PNH, C3G or primary IC-MPGN.
Elderly population.
Although there were no apparent age-related differences observed in these studies, the number of patients aged 65 years and over was not sufficient to determine whether they respond differently from younger patients.
Paediatric population.
Based on population pharmacokinetic analysis, body weight in adolescent patients (12-17 years) has an impact on clearance and volume of distribution. The dosing regimen for adolescents with C3G or primary IC-MPGN is based on the patient's body weight. The model-predicted exposure for adolescents with C3G or primary IC-MPGN is adequately matched to the adult reference exposure.
Renal insufficiency.
In a study of 8 patients with severe renal impairment, defined as creatine clearance (CrCl) less than 30 mL/min (with 4 patients with values less than 20 mL/min), renal impairment had no effect on the pharmacokinetics of pegcetacoplan (see Section 4.2 Dose and Method of Administration). Based on population pharmacokinetic analysis, eGFR had no clinically meaningful impact on pegcetacoplan exposure in a pooled analysis population.
5.3 Preclinical Safety Data
Genotoxicity.
Pegcetacoplan was not mutagenic when tested in in vitro bacterial reverse mutation (Ames) assays and was not clastogenic in an in vitro assay in human TK6 cells or in an in vivo micronucleus assay in mice.
Carcinogenicity.
Long-term animal carcinogenicity studies of pegcetacoplan have not been conducted.6 Pharmaceutical Particulars
6.1 List of Excipients
Sorbitol, glacial acetic acid, sodium acetate trihydrate, sodium hydroxide, water for injections.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store at 2°C - 8°C in a refrigerator. Do not freeze.
Keep Empaveli in its original package to protect from light.
6.5 Nature and Contents of Container
Sterile solution present in a vial (Type I glass) with a stopper (chlorobutyl or bromobutyl), and a seal (aluminium) with a flip-off cap (polypropylene).
Each 20 mL vial contains 1.3 mL overfill.
Pack sizes of 1 or 8 vials. Not all pack sizes may be marketed.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Pegcetacoplan, the active ingredient in Empaveli solution for subcutaneous infusion 1,080 mg/20 mL, is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to both ends of a linear polyethylene glycol (PEG) molecule. The molecular weight of pegcetacoplan is approximately 43.5 kiloDaltons (kDa). The peptide moieties bind to complement C3 and exert a broad inhibition of the complement cascade. The 40-kDa PEG moiety imparts improved solubility and longer residence time in the body after administration of the drug product. The structure of pegcetacoplan is shown below.
Chemical structure.
 The chemical name is poly(oxy-1,2-ethanediyl), α-hydro, ω-hydroxy-,15,15'-diester with N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-α-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-[2-(2-aminoethoxy)ethoxy]acetyl-N6-carboxy-L-lysinamide cyclic (2→12)-(disulfide).
The chemical name is poly(oxy-1,2-ethanediyl), α-hydro, ω-hydroxy-,15,15'-diester with N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-α-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-[2-(2-aminoethoxy)ethoxy]acetyl-N6-carboxy-L-lysinamide cyclic (2→12)-(disulfide).
The chemical formula is C170H248N50O47S4.(C2H4O)n n = 800-1100.
CAS registry number.
2019171-69-6.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
