1 Name of Medicine
Enrylaze (recombinant crisantaspase).
2 Qualitative and Quantitative Composition
Each vial of 0.5 mL solution contains 10 mg of crisantaspase (L-asparaginase from Erwinia chrysanthemi) produced by recombinant DNA technology in Pseudomonas fluorescens.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection/infusion.
Clear to opalescent, colourless to slightly yellow solution.
4.1 Therapeutic Indications
Enrylaze is indicated as a component of a multi-agent chemotherapeutic regimen, for the treatment of acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma (LBL) in adults and paediatric patients (1 month and older) who have developed hypersensitivity or silent inactivation to E. coli-derived asparaginase.
4.2 Dose and Method of Administration
Administer Enrylaze in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis.
Administer premedication (paracetamol, an H1 receptor blocker, and an H2 receptor blocker) 30-60 minutes prior to Enrylaze to decrease the risk and severity of hypersensitivity reaction.
Dosage.
Enrylaze is dosed in mg/m2 and not in units/m2 as used for other asparaginase preparations. Crisantaspase products may not be bioequivalent and should not be assumed to be interchangeable.
The recommended dosage of Enrylaze follows either a Monday/Wednesday/Friday schedule, or a 48-hourly schedule:
Monday/Wednesday/Friday: 25 mg/m2 on Mondays; 25 mg/m2 on Wednesdays; 50 mg/m2 on Fridays.
48-hourly: 25 mg/m2 once every 48 hours.
Each dose can be given either intramuscularly (IM) or intravenously (IV). IV administration may be less painful at the site of administration but it results in serum asparaginase activity (SAA) with a higher initial peak (Cmax) and a lower trough prior to the next dose compared to IM administration (see Section 5.2 Pharmacokinetic Properties). It may also have a higher risk of some toxicities including hypersensitivity reaction (see Section 4.4 Special Warnings and Precautions for Use).
Monitor nadir (pre-dose/trough) SAA (NSAA) and adjust dosing in accordance with local treatment protocols. An NSAA of < 0.1 U/mL is associated with loss of efficacy. Particularly, if using the Monday/Wednesday/Friday dosing schedule, and the Friday dose is administered IV, assess NSAA just prior to the Monday dose. NSAA may be increased by reducing the number of hours between doses (by adjusting dose time of day), or by changing to a 48-hourly schedule (if on Monday/Wednesday/Friday dosing), or by changing from IV to IM administration.
The 48-hourly dosing schedule is based on modelling and simulations, without direct/observed clinical data.
Dosage adjustment.
Hepatic impairment.
Dose adjustment is not required for patients that develop total bilirubin that remains less than 3 times the Upper Limit of Normal (ULN) during treatment.
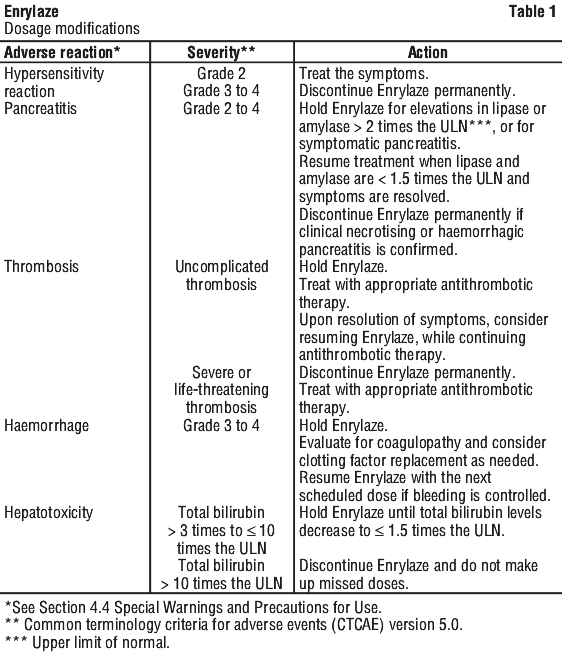
If bilirubin elevation 3 times the ULN occurs during treatment, modify treatment according to Table 1.
Renal impairment.
There are insufficient data in patients with mild, moderate or severe renal impairment to support a dose recommendation.
Paediatric population.
No dose adjustment is required in paediatric patients.
The safety and efficacy of children aged younger than 1 month has not yet been established.
Elderly.
No dose adjustment is required in elderly patients.
Adverse reactions.
If an adverse reaction occurs, modify treatment according to Table 1.
Method of administration.
Product is for single use in one patient only. Discard any residue.
Enrylaze is given either by intramuscular (IM) or intravenous (IV) administration, and the mode of administration changes its pharmacokinetics (see Dosage, above; Section 5.2 Pharmacokinetic Properties).
For IM use, limit the volume of Enrylaze at a single injection site to 1 mL (for patients with a body surface area (BSA) < 0.5 m2), or 2 mL (for patients with a BSA ≥ 0.5 m2). If the volume to be administered is greater than this limit, use multiple injection sites.
For IV infusion, it is recommended to administer the dose over 2 hours.
Compatibilities.
Do not mix Enrylaze with other medicinal products prior to administration.
Enrylaze is compatible with administration using the following materials (no other materials have been studied):
Syringes made of polypropylene.
Intravenous infusion sets made of PVC, polyolefin, polyamide, and ethylene vinyl acetate.
Preparation instructions.
Determine the posology, and number of vials of Enrylaze based on the individual patient's BSA as outlined above. More than one vial may be needed for a full dose.
Remove the appropriate number of vials of Enrylaze from the refrigerator.
Do not shake the vial.
Each vial should be inspected for particles. If particles are observed and/or the liquid in the vial is not clear, the vial must not be used.
Withdraw the required volume of Enrylaze into a syringe.
If the prepared dose is not used immediately the syringe containing the prepared dose can be stored at room temperature for 8 hours or in a refrigerator (2°C to 8°C) for up to 24 hours (see Section 6.3 Shelf Life).
Subsequent steps for IV infusion preparation.
The prepared dose of Enrylaze in the syringe should be further diluted in an infusion bag containing 100 mL of sodium chloride 9 mg/mL (0.9%).
The IV infusion prepared dose should be a clear liquid free from visual particulates.
If particles are observed in the IV infusion prepared dose the solution must not be used.
If the IV infusion prepared dose is not used immediately, it can be stored at room temperature (15°C-25°C) for 12 hours or refrigerated (2°C to 8°C) for up to 24 hours. The start of storage mentioned starts from withdrawing the required volume from the vial (see Section 6.3 Shelf Life).
The 12 or 24-hour storage time includes the recommended 2-hour infusion time.4.3 Contraindications
History of severe hypersensitivity reactions to the active substance.
Hypersensitivity to any of the excipients listed in Section 6.1 List of Excipients.
Severe pancreatitis.
History of severe pancreatitis during previous asparaginase therapy.
Severe thrombosis during previous asparaginase therapy.
Severe haemorrhagic events during previous asparaginase therapy.
4.4 Special Warnings and Precautions for Use
Enrylaze treatment should be prescribed by physicians and administered by health care personnel experienced in the use of antineoplastic products and in the treatment of haematological malignancies.
To enable linking of exposed patients to batch numbers, the tradename and batch number of the administered product should be clearly recorded in the patient file.
Clinical monitoring.
The effect of Enrylaze on serum asparaginase activity (SAA) varies between patients. Monitor nadir/trough SAA according to local treatment protocols and adjust dosage as needed (see Section 4.2 Dose and Method of Administration).
Hypersensitivity reactions.
Hypersensitivity reactions were very common amongst patients receiving Enrylaze in clinical trials (see Section 4.8 Adverse Effects (Undesirable Effects); Section 4.3 Contraindications). The risk may be higher with intravenous than intramuscular administration. Across both routes of administration, severe events occurred in 8% of patients, including anaphylaxis in 2% of patients. Drug hypersensitivity led to discontinuation for 10% of patients.
Administer Enrylaze in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis. Discontinue Enrylaze for severe hypersensitivity reactions (see Section 4.2 Dose and Method of Administration, Table 1).
Pancreatitis.
Pancreatitis was reported in 7% of patients who received Enrylaze in clinical trials. The incidence of serious events was 5% and of life-threatening events was 1%. Pancreatitis led to treatment discontinuation in 5% of patients in study JZP458-201.
Inform patients of the signs and symptoms of pancreatitis, and that if left untreated pancreatitis can be fatal. Withhold or discontinue Enrylaze for pancreatitis based on severity (see Section 4.2 Dose and Method of Administration, Table 1).
Thrombosis.
Thrombosis was reported in 2% of patients and led to a discontinuation of treatment for one patient. Serious thrombotic events including sagittal sinus thrombosis and pulmonary embolism were reported in 1% of patients who received Enrylaze in clinical trials. Withhold Enrylaze for a thrombotic event and administer appropriate antithrombotic therapy. Consider resumption of treatment with Enrylaze only if the patient had an uncomplicated thrombosis (see Section 4.2 Dose and Method of Administration, Table 1).
Haemorrhage.
Haemorrhage was reported in 18% of patients who received Enrylaze in clinical trials; severe haemorrhage occurred in 2% of patients. The most commonly reported haemorrhage-related events were contusion (10%), epistaxis (8%), petechiae (3%) and menorrhagia (1%). In patients treated with L-asparaginase class products, haemorrhage may be associated with increased prothrombin time (PT), increased partial thromboplastin time (PTT), and hypofibrinogenaemia. Consider appropriate replacement therapy in patients with severe or symptomatic coagulopathy (see Section 4.2 Dose and Method of Administration, Table 1).
Hepatotoxicity.
Elevated transaminases occurred in 29% of patients, of which 17% were Grade 3 or higher. Elevated blood bilirubin occurred in 10% of patients, of which 2% were Grade 3 or higher amongst patients who received Enrylaze in clinical trials (see Section 4.8 Adverse Effects (Undesirable Effects)). Asparaginase hepatotoxicity typically involves rapid onset steatosis and jaundice.
Hepatic veno-occlusive disease (VOD), which can be fatal, can also occur with asparaginase-containing chemotherapeutic regimens, though it is more common with pegaspargase than L-asparaginases. Hepatic VOD may present with rapid weight gain, fluid retention with ascites, hepatomegaly (which may be painful), rapid increase of bilirubin, refractory thrombocytopenia and multiorgan dysfunction.
Inform patients of the signs and symptoms of hepatotoxicity. Evaluate bilirubin and transaminases prior to each cycle of Enrylaze and at least weekly during cycles of treatment that include Enrylaze, through four weeks after the last dose of Enrylaze. For patients who develop abnormal liver tests during treatment with Enrylaze, increase the frequency of monitoring. In the event of serious liver toxicity, discontinue treatment with Enrylaze and provide supportive care (see Section 4.2 Dose and Method of Administration, Table 1).
Glucose intolerance.
Cases of glucose intolerance have been reported in patients receiving Enrylaze in clinical trials (see Section 4.8 Adverse Effects (Undesirable Effects)). Monitor glucose levels in patients at baseline and periodically during treatment. Administer insulin therapy as necessary.
Use in the elderly.
No data available.
Paediatric use.
No data is available in patients under 1 month of age.
Effects on laboratory tests.
See Section 4.8 Adverse Effects (Undesirable Effects).4.5 Interactions with Other Medicines and Other Forms of Interactions
General.
No dedicated interaction studies have been performed.
As Enrylaze may impair liver function and decrease protein synthesis, it may interact with other medicines whose pharmacokinetics or pharmacodynamics are affected by changes in liver function or plasma protein levels.
Vincristine.
Administration of asparaginase concurrently or immediately before vincristine may be associated with increased toxicity of vincristine. Asparaginase inhibits hepatic clearance of vincristine.
Methotrexate, cytarabine.
Non-clinical data indicates that prior or concurrent administration of L-asparaginase attenuates the effect of methotrexate and cytarabine. Administration of L-asparaginase after methotrexate or cytarabine results in a synergistic effect. However, the clinical effect of sequence-dependent L-asparaginase administration on the efficacy of methotrexate and cytarabine is unknown.
Glucocorticoids.
Administration of asparaginase with or immediately before glucocorticoids (e.g. prednisone) may change coagulation parameters, such as a decrease in fibrinogen and antithrombin III levels.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no clinical data on the effect of recombinant crisantaspase on fertility.
In a fertility and early embryonic development study in rats with Erwinia chrysanthemi crisantaspase, there were no effects on female or male fertility with repeat doses up to 12,000 IU/m2 administered intramuscularly (margins of human exposure < 1). Sperm counts were slightly decreased in males at ≥ 3000 IU/m2 given every other day for 10 weeks.
All patients should use highly effective contraception (other than oral contraceptives) during treatment with Enrylaze and for at least 3 months after discontinuation.
Oral contraceptives could be ineffective, because an indirect interaction between oral contraceptives and Enrylaze has not been ruled out.
(Category D)
There are no clinical data on the use of recombinant crisantaspase in pregnancy. Based on its mechanism of action, and studies with Erwinia chrysanthemi crisantaspase in pregnant animals, recombinant crisantaspase can cause embryonic and fetal harm when administered during pregnancy. Advise patients of the potential risk to a fetus, and verify pregnancy status before initiation of Enrylaze.
In non-clinical studies with Erwinia chrysanthemi crisantaspase the following was observed:
In embryofetal development studies in rats and rabbits, Erwinia chrysanthemi crisantaspase produced maternal toxicity, increased resorptions, post implantation loss, embryofetal toxicity, and/or gross abnormalities with intramuscular doses of 12,000 IU/m2 in rat and 480 IU/m2 in rabbit (margins of exposure < 1). Findings in rats included increased incidence of partially undescended thymic tissue in the fetus and decreased maternal body weight gain. Findings in rabbits were decreased maternal body weight gain, increased resorption and post-implantation loss and associated decrease in the number of live fetuses, and gross abnormalities (e.g. absent kidney, absent accessory lung lobe, additional subclavian artery, and delayed ossification).
In a pre- and postnatal development study in rats there were no adverse effects on gestation, parturition, or growth, development or reproductive performance of offspring following intramuscular doses to the dam at 14,400 IU/m2 (margins of exposure < 1) every other day from gestation day 6 to postnatal day 20.
It is not known whether recombinant crisantaspase is excreted in human milk. Because of the potential for serious adverse reactions in breastfeeding infants/children, advise patients not to breastfeed during Enrylaze therapy and for a period of two weeks after the last dose.4.7 Effects on Ability to Drive and Use Machines
Based on the adverse reactions of Enrylaze there may be a minor influence on the ability to drive and use machines (see Section 4.8 Adverse Effects (Undesirable Effects)).
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
Serious adverse reactions occurred in 59% of patients who received Enrylaze in a clinical trial. The most frequent serious adverse reactions (in ≥ 5% of patients) were febrile neutropenia, pyrexia, vomiting, sepsis, drug hypersensitivity, nausea and pancreatitis.
The most common adverse reactions (in ≥ 20% patients) were anaemia, vomiting, thrombocytopenia, neutropenia, nausea, febrile neutropenia, fatigue, pyrexia, decreased appetite, transaminase increased, abdominal pain, white blood cell count decreased, headache, diarrhoea and lymphocyte count decreased.
Tabulated list of adverse reactions.
The safety of Enrylaze was evaluated in Study JZP458-201, an open-label, multi-cohort, multi-centre study in which 228 patients with acute lymphoblastic leukaemia (ALL) or lymphoblastic lymphoma (LBL) who had developed hypersensitivity to a long-acting E. coli-derived asparaginase received Enrylaze as a component of multi-agent chemotherapy (see Section 5.1 Pharmacodynamic Properties, Clinical trials). All of the studied regimens involved Monday/Wednesday/Friday dosing schedules, so there is no direct clinical safety data for the 48-hourly dosing regimen.
The most common adverse effects reported with the use of Enrylaze as a component of multi-agent chemotherapy in Study JZP458-201 are summarised in Table 2.
Certain ADRs listed (see Table 2), such as reactions resulting from bone marrow suppression, and infections, are known to be associated with multi-agent chemotherapeutic regimens, and the contributory role of Enrylaze is not clear. In individual cases of adverse reactions, other medicinal products of the regimen may have contributed.
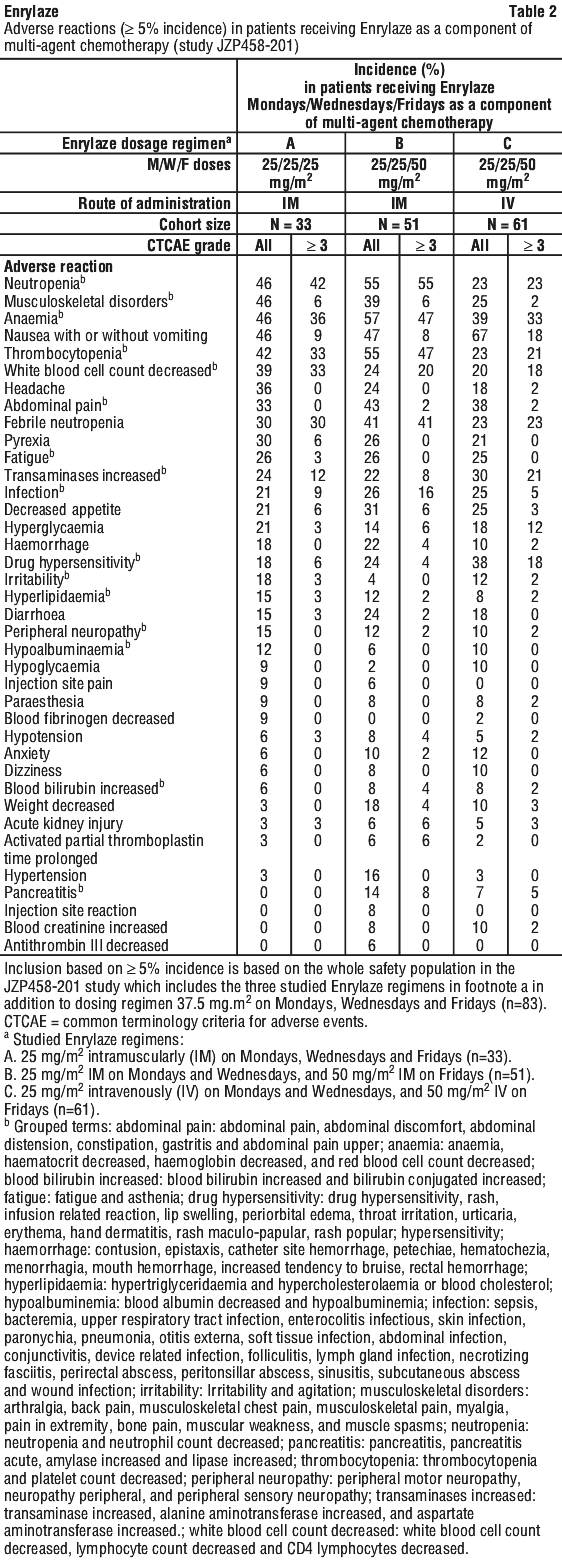 Clinically relevant adverse reactions in < 5% of patients who received Enrylaze in combination with chemotherapy included: acute respiratory distress syndrome (0.4%); acidosis (0.9%); anaphylaxis (2%); gait disturbance (4%); hyperammonaemia (2%) including 1 case of hyperammonaemic encephalopathy (0.4%); pulmonary oedema (1%); and thrombosis/thromboembolic events (total 2%: including, jugular vein thrombosis (0.9%), deep vein thrombosis (0.4%), superior sagittal sinus thrombosis (0.4%) and pulmonary embolism (1%).
Clinically relevant adverse reactions in < 5% of patients who received Enrylaze in combination with chemotherapy included: acute respiratory distress syndrome (0.4%); acidosis (0.9%); anaphylaxis (2%); gait disturbance (4%); hyperammonaemia (2%) including 1 case of hyperammonaemic encephalopathy (0.4%); pulmonary oedema (1%); and thrombosis/thromboembolic events (total 2%: including, jugular vein thrombosis (0.9%), deep vein thrombosis (0.4%), superior sagittal sinus thrombosis (0.4%) and pulmonary embolism (1%).
Postmarketing experience.
The following adverse events have been reported during post approval use of crisantaspase. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Hepatic.
Veno-occlusive disease (VOD).
Paediatric population.
The majority of the patients in Study JZP458-201 (86%) were paediatric patients (under the age of 18 years).
Adults and other special populations.
Enrylaze has not been studied in patients older than 25 years. Some adverse reactions, such as hepatotoxicity, thrombosis, and pancreatitis, have been reported more frequently in adults with acute lymphoblastic leukemia receiving other asparaginases than in paediatric patients.
Immunogenicity.
It has been reported that there is no to little cross reactivity between crisantaspase and other E. coli derived asparaginase. As with all therapeutic proteins, there is a potential for immunogenicity. The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay and may be influenced by several factors such as assay methodology, sample handling, timing of sample collection, concomitant treatment, and underlying disease. Differences in assay methods preclude meaningful comparison between studies.
During treatment in Study JZP458-201 (range 1 to 15 courses), 108 of 228 (47%) patients receiving Enrylaze (by either intramuscular injection (n=167) or intravenous infusion (n=61)) developed treatment-emergent anti-drug antibodies (ADA) against Enrylaze; a further 8 patients (4%) were also ADA-positive but already had ADA present prior to their first dose.
A total of 23 (20%) patients who had ADAs experienced hypersensitivity reactions of which 6 (5%) had neutralising antibodies. Of the patients without any ADAs, 11/112 (10%) experienced a hypersensitivity reaction.
During the course of treatment 73 (63%) patients became ADA negative at least once.
Intravenous infusion.
A total of 34 (56%) patients were found to be ADA positive.
1 patient was ADA positive at pre dose 1.
33 patients developed ADA toward Enrylaze following administration of Enrylaze. 18 of these patients subsequently became ADA negative at least once during the study.
12 (35%) experienced hypersensitivity reactions during the study, and of these patients 2 had neutralising antibodies. Of the negative ADA patients 4/27 (15%) experienced a hypersensitivity reaction.
Intramuscular injection.
A total of 82 (49%) patients were found to be ADA positive.
7 patients were ADA positive at pre dose 1.
75 patients developed ADA toward Enrylaze following administration of Enrylaze. 55 of these patients subsequently became ADA negative at least once during the study.
11 (13%) patients experienced hypersensitivity reactions, and of these patients 4 had neutralising antibodies. Of the negative ADA positive patients 7/85 (8%) experienced a hypersensitivity reaction.
The presence of ADA does not appear to correlate with the occurrence of hypersensitivity reactions. SAA levels were not impacted for applicable ADA positive patients as they maintained SAA levels ≥ 0.1 U/mL at all available 48- and 72-hour time points during Course 1. No impact on the pharmacokinetics of Enrylaze was observed and ADA status was not found to be a significant factor in population pharmacokinetic analysis.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product.
Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
No case of Enrylaze overdose with clinical symptoms has been reported and there is no specific antidote. Symptomatic and supportive treatment is recommended.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: other antineoplastic agents ATC code: L01XX02.
Mechanism of action.
Asparaginase is an enzyme that catalyses the conversion of the amino acid L-asparagine into L-aspartic acid and ammonia. The pharmacological effect of Enrylaze is based on the killing of leukaemic cells due to depletion of plasma asparagine. Leukaemic cells with low expression of asparagine synthetase have a reduced ability to synthesise asparagine,and therefore are dependent on an exogenous source of asparagine for survival.
Serum asparaginase activity.
An in vitro activity assay demonstrated that 1 mg of recombinant crisantaspase approximates 1,000 U of native crisantaspase, consistent with the in vivo comparisons from clinical trials. Serum asparaginase activity (SAA) exposures (Cmax, concentration at 48h and 72h and AUC) have been shown to be comparable for 25 mg/m2 recombinant crisantaspase and 25 000 U/m2 native crisantaspase, when administered intravenously or intramuscularly in healthy subjects.
Clinical trials.
The efficacy and safety of Enrylaze as a component of a multi-agent chemotherapeutic regimen for the treatment of patients with ALL or LBL who have developed hypersensitivity to E. coli-derived asparaginase was evaluated in Study JZP458-201, an open-label, multi-cohort, multi-centre trial. In this trial, in substitution for pegaspargase (usually given once per fortnight), Enrylaze was administered at various dosages and routes of administration every Monday, Wednesday, and Friday for a total of six doses each fortnight.
The studied Enrylaze regimens were:
25 mg/m2 intramuscularly (IM) on Mondays, Wednesdays and Fridays (n=33).
37.5 mg/m2 IM on Mondays, Wednesdays and Fridays (n=83); not an approved regimen.
25 mg/m2 IM on Mondays and Wednesdays, and 50 mg/m2 IM on Fridays (n=51).
25 mg/m2 intravenously (IV) on Mondays and Wednesdays, and 50 mg/m2 IV on Fridays (n=61).
The median age of patients was 10 years (range, 1 to 25 years); 61% of patients were male, 69% were Caucasian, 83% of patients had experienced a hypersensitivity reaction (Grade ≥ 3) to pegaspargase, 7% had experienced silent inactivation, and 10% had experienced an allergic reaction with inactivation. The number of six-dose courses of Enrylaze received ranged from 1 to 15.
Efficacy was established based on the demonstration of achievement and maintenance of nadir serum asparaginase activity (NSAA) levels of at least 0.1 IU/mL. Serum trough asparaginase activity ≥ 0.1 IU/mL correlates with asparagine depletion that predicts clinical efficacy (see Section 5.2 Pharmacokinetic Properties). Observed NSAA levels in Study JZP458-201 for approved dosing schedules are presented in Table 3.
 The other recommended dosing schedules are based on interpolation from population pharmacokinetic (PK) modelling and simulation. Also see Section 5.2 Pharmacokinetic Properties.
The other recommended dosing schedules are based on interpolation from population pharmacokinetic (PK) modelling and simulation. Also see Section 5.2 Pharmacokinetic Properties.
Paediatric population.
No clinically significant difference is expected in probability of achieving a therapeutic NSAA ≥ 0.1 U/mL based on age (1 month to 39 years) when using the recommended body surface area (BSA)-based dosing regimens.
Safety and efficacy for patients under the age of 1 month have not been determined.
5.2 Pharmacokinetic Properties
The pharmacokinetics (PK) of Enrylaze was determined based on serum asparaginase activity (SAA). Patients with ALL or LBL received Enrylaze in Study JZP458-201 at various dosages intramuscularly or intravenously (see Section 5.1 Pharmacodynamic Properties, Clinical trials) as a replacement for each dose of a long-acting E. coli-derived asparaginase, whilst otherwise remaining on their original treatment plan. Recombinant crisantaspase maximum SAA (Cmax) and area under the SAA-time curve (AUC) increase proportionally over a dosage range from 12.5 to 50 mg/m2. The exposures for recombinant crisantaspase are summarised in Table 4.

Absorption.
The median Tmax of recombinant crisantaspase is 16 Fhours following IM administration. The mean absolute bioavailability for IM administration is 38% in healthy subjects.
Distribution.
Following IV administration, the geometric mean (%CV) volume of distribution of recombinant crisantaspase is 1.75 L/m2 (14%).
Metabolism.
Recombinant crisantaspase is expected to be metabolised into small peptides by catabolic pathways.
Excretion.
Following IV administration, the geometric mean (%CV) clearance of recombinant crisantaspase is 0.14 L/hour/m2 (20%).
The geometric mean (%CV) half-life is 8.6 hours (13%) following IV administration and 18.8 hours (11%) following IM administration.
Special populations.
Renal and hepatic impairment.
There was no dedicated study on renal or hepatic impairment with Enrylaze.
During treatment dose adjustment is not required for patients with total bilirubin ≤ 3 times the Upper Limit of Normal; there is limited data with Enrylaze in patients with total bilirubin > 3 times to ≤ 10 times the ULN.
Age, weight, body surface area and sex.
There were no clinically significant differences in the pharmacokinetics of Enrylaze based on weight (9 to 131 kg) or sex (n=138 male; n=88 female) after the dose was adjusted by body surface area (BSA).
The volume of distribution and clearance of recombinant crisantaspase increase with increasing BSA (0.44 to 2.53 m2).
Age impacts absorption rate constant whereas younger subjects have higher absorption rate constant value, leading to earlier Tmax.
Race.
Black or African American patients (n=24) had 25% lower clearance which may increase SAA exposure compared to population average (n=226). No dose adjustment is needed in African American population.
Neutralising antibodies.
As with other asparaginase preparations, development of specific neutralising antibodies were identified with repeated dosing. See Section 4.8 Adverse Effects (Undesirable Effects), Immunogenicity.
5.3 Preclinical Safety Data
Genotoxicity.
No data available. Genotoxicity of crisantaspase was not investigated, but crisantaspase is not expected to be genotoxic.
Carcinogenicity.
No data available. No long-term carcinogenicity studies in animals have been performed with crisantaspase.6 Pharmaceutical Particulars
6.1 List of Excipients
Trehalose dihydrate, sodium chloride, sodium hydroxide (pH adjustment), dibasic sodium phosphate, monobasic sodium phosphate monohydrate, polysorbate 80, water for injections.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products. This includes infusion of other medicinal products using the same infusion line as Enrylaze.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
In-use stability data.
Intramuscular preparation.
Chemical and physical in-use stability for IM preparations in a polypropylene syringe has been demonstrated for up to 8 hours at room temperature (15°C-25°C) or 24 hours when refrigerated (2°C-8°C).
Intravenous preparation.
Chemical and physical in-use stability for IV preparations has been demonstrated for up to 12 hours at room temperature (15°C-25°C) or 24 hours when refrigerated (2°C-8°C).
The storage times start from withdrawing the required volume from the unopened vials.
The storage time in the polyethylene inner lined IV bag includes the 2-hour administration time (see Section 4.2 Dose and Method of Administration).
6.4 Special Precautions for Storage
Store in a refrigerator (2°C-8°C) in an upright position. Keep the vial in the outer carton in order to protect from light.
Do not shake or freeze.
For in-use storage conditions of the medicinal product, see Section 6.3 Shelf Life.
6.5 Nature and Contents of Container
Type 1 clear borosilicate glass vial with a capacity of 2 mL sealed with a halobutyl rubber stopper and aluminium overseal and a violet plastic cap.
Pack size: 3 vials.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.

CAS number.
CAS registry number: 1349719-22-7.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.