What is in this leaflet
This leaflet answers some common questions about Entyvio.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available. You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on this medicine. You can also download the most up to date leaflet from https://www.takeda.com/en-au.
Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given this medicine against the benefits they expect it will have for you.
If you have any concerns about being given this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Entyvio is used for
Entyvio contains the active substance vedolizumab, a monoclonal antibody. Monoclonal antibodies are proteins that recognise and bind to certain special proteins in the body.
Entyvio specifically binds to a protein called integrin α 4 β 7 present on certain white blood cells. Integrin α 4 β 7 can act to increase inflammation seen in ulcerative colitis and Crohn’s disease.
Entyvio works by blocking α 4 β 7 integrins and so reduces inflammation.
Ulcerative colitis
Ulcerative colitis is an inflammatory disease of the large bowel. Entyvio is used to treat the signs and symptoms of moderate to severe ulcerative colitis in adults who have not responded well enough or are intolerant to other treatments.
Crohn’s disease
Crohn’s disease is an inflammatory disease of the bowel. It may also affect any part of the gut. Entyvio is used to treat the signs and symptoms of moderate to severe Crohn’s disease in adults who have not responded well enough or are intolerant to other treatments.
Ask your doctor or pharmacist if you have any questions about why Entyvio has been prescribed for you. Your doctor may have prescribed it for another purpose.
This medicine is available only with a doctor’s prescription.
There is not enough information to recommend the use of this medicine for children under the age of 18 years.
Before you are given Entyvio
When you must not be given it
You should not be given Entyvio if you:
- are allergic to vedolizumab or any of the other ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction may include wheezing, difficulty breathing, hives, itching, swelling or dizziness. - have an active severe infection, such as for example tuberculosis, blood poisoning, serious abscesses.
Do not receive it after the expiry date printed on the pack or if the packaging is damaged or shows signs of tampering. If it has expired or is damaged return it to your pharmacist for disposal.
Before you are given it
Tell your doctor if:
- you have an infection, or think you have an infection.
- are going to receive any vaccination or have recently had a vaccination.
Entyvio may affect the way you respond to a vaccination - you have any allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you are pregnant or intend to become pregnant. The effects of Entyvio in pregnant women are not known. Therefore, this medicine is not recommended for use during pregnancy unless you and your doctor decide that the benefit to you clearly outweighs the potential risk to your baby.
Your doctor can discuss the risks and benefits involved.
If you are a woman of childbearing potential, you should use adequate contraception to avoid falling pregnant and continue to use it for at least 4.5 months after the last treatment with Entyvio.
Tell your doctor if you are breastfeeding or intend to breastfeed. Entyvio passes into breast milk and it is not known what effect this may have on your baby.
Women who are breastfeeding or intend to breastfeed should talk to their doctor about whether or not to use Entyvio.
If you have not told your doctor about any of the above, tell them before you are given Entyvio.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food store.
Entyvio should not be given with other biologic medicines that suppress your immune system as this combination has not been studied in clinical trials.
If you have previously taken natalizumab (TYSABRI) or rituximab (MABTHERA) for other conditions, tell your doctor who will decide if you can be given Entyvio.
How Entyvio is given
Entyvio will be given to you by your doctor or nurse, through a drip in one of the veins in your arm (intravenous infusion) over about 30 minutes.
Your healthcare provider will monitor you during and after the Entyvio infusion for side effects to see if you have a reaction to the treatment.
If you have any questions about how this medicine is given, ask your doctor or nurse who is giving you the infusion.
How much to take
Treatment with Entyvio is the same for ulcerative colitis and Crohn’s disease.
The recommended dose of Entyvio is 300 mg per infusion. Following the initial dose, you will get additional infusions at 2 and 6 weeks and be assessed by your treating doctor 6 to 8 weeks after you’ve completed the initial treatment doses to see how you have responded. For patients with Crohn’s Disease who have not shown response, your doctor may recommend an additional dose of Entyvio at week 10.
If there has been an adequate response to the first 3 doses you may continue treatment with further doses every 8 weeks. Your doctor may decide to alter this treatment schedule to every 4 weeks depending on how well Entyvio works for you.
How long to take it
Continue receiving your medicine for as long as your doctor tells you. You should not stop using Entyvio without talking with your doctor first.
If you forget or miss an appointment
If you forget or miss an appointment to receive the infusion, make another appointment as soon as possible.
If you are not sure what to do, ask your doctor.
If you are given too much (overdose)
As Entyvio is given to you by infusion under the supervision of a doctor or nurse, it is unlikely that you will receive too much.
However, if you experience any side effects after being given Entyvio, tell your doctor or nurse immediately.
While you are being given Entyvio
Things you must do
When you first receive this medicine and during the course of the treatment, also between doses, tell your doctor or nurse immediately if you:
- experience blurred, loss of or double vision, difficulty speaking, weakness in an arm or a leg, a change in the way you walk or problems with your balance, persistent numbness, decreased sensation or loss of sensation, or memory loss or confusion. These may all be symptoms of a serious and potentially fatal brain condition known as progressive multifocal leukoencephalopathy (PML).
Your doctor or nurse will provide you with a Patient Alert Card, that you should keep with you at all times. This card will remind you to tell all other health professionals that you are being given Entyvio.
If you experience any of the symptoms in this card, you should contact your doctor immediately. - have an infection, or think you have an infection, such as if you develop chills, shivering, persistent cough or a high fever.
Some infections may become serious and possibly even life-threatening if left untreated - experience signs of an allergic reaction or other reaction to the infusion such as wheezing, difficulty breathing, hives, itching, swelling or dizziness.
These could occur during or after the infusion. - are going to receive any vaccination or have recently had a vaccination.
Entyvio may affect the way you respond to a vaccination
If you are about to be started on any new medicine, remind your doctor that you are taking Entyvio.
If you become pregnant while you are taking this medicine, tell your doctor immediately.
Things you must not do
Do not use this medicine to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not stop taking Entyvio without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how Entyvio affects you. This medicine may have a minor influence on your ability to drive or use tools or machines. A small number of patients have felt dizzy after receiving Entyvio.
If you feel dizzy, do not drive or use tools or machines.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Entyvio.
Entyvio helps most people with ulcerative colitis or Crohn’s disease, but it may have unwanted side effects in a few people.
All medicines have some unwanted side effects. Sometimes they are serious, but most of the time they are not. You may need medical attention if you get some of the side-effects.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or nurse if you notice any of the following and they worry you:
- headache
- fever
- common cold
- flu (influenza)
- nose or throat infection
- bronchitis
- chest infection
- cough
- throat pain
- nausea
- itching
- rash and redness
- pain in the extremities
- back pain
- joint pain
- tiredness
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor as soon as possible if you notice any of the following:
- high fever
- rash
- dark urine, yellowing of the eyes and skin (jaundice)
- severe upper stomach pain, often with nausea and vomiting or stomach pain that spreads to your back.
The above symptoms could be a sign of infection, liver injury or inflammation of the pancreas. Some infections may become serious and possibly even life-threatening if left untreated.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- wheezing or difficulty breathing
- hives
- itching of the skin
- swelling
- rapid heart beat
- vomiting
- chills or shivering
The above list includes very serious side effects. You may need urgent medical attention. These side effects are rare.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may occur in some people.
Storage
If you need to store Entyvio before taking it to hospital or clinic, the unopened vial should be stored in the refrigerator at 2°C to 8°C (Refrigerate. Do not freeze). Protect from light. Keep the vial in the original carton.
Do not store it or any other medicine in the bathroom, near a sink, or on a windowsill.
Do not leave it in the car. Heat and damp can destroy some medicines.
Keep it where children cannot reach it.
Product description
What it looks like
Entyvio is a white to off-white powder provided in a glass vial.
Each pack of Entyvio contains one vial.
Ingredients
Active ingredient:
Each vial of Entyvio contains 300 mg of vedolizumab.
After reconstitution each mL of solution contains 60 mg of vedolizumab.
Other ingredients:
- histidine
- histidine hydrochloride monohydrate
- arginine hydrochloride
- sucrose
- polysorbate 80
Sponsor
Entyvio is supplied in Australia by:
Takeda Pharmaceuticals
Australia Pty Ltd
Level 39, 225 George Street
Sydney, NSW 2000
Australia
Tel: 1800 012 612
www.takeda.com/en-au
Entyvio is supplied in New Zealand by:
Takeda New Zealand Limited
Level 10, 21 Queen Street
Auckland 1010
New Zealand
Tel: 0508 169 077
This leaflet was prepared in September 2022.
Australian Registration Number
AUST R 210048
ENTYVIO® and the ENTYVIO Logo® are registered trademarks of Millennium Pharmaceuticals, Inc., a Takeda company. TAKEDA® and the TAKEDA Logo® are registered trademarks of Takeda Pharmaceutical Company Limited.
Published by MIMS September 2023
 No clinically relevant differences in the overall safety profile and adverse events were observed in patients who received vedolizumab in the EARNEST trial (pouchitis). Rectal bleeding, asthenia and chest discomfort were reported in vedolizumab group (3%).
No clinically relevant differences in the overall safety profile and adverse events were observed in patients who received vedolizumab in the EARNEST trial (pouchitis). Rectal bleeding, asthenia and chest discomfort were reported in vedolizumab group (3%). In exploratory analyses, the beneficial effect of intravenous vedolizumab on clinical response, remission and mucosal healing was observed both in patients with no prior TNFα antagonist exposure and in those who had failed prior TNFα antagonist therapy.
In exploratory analyses, the beneficial effect of intravenous vedolizumab on clinical response, remission and mucosal healing was observed both in patients with no prior TNFα antagonist exposure and in those who had failed prior TNFα antagonist therapy. In the GEMINI I study, the induction regimen was administered at weeks 0 and 2 and maintenance dosing started at week 6. However exploratory analyses suggest a higher rate of long term clinical response and remission will be achieved with a 0, 2 and 6 week induction regimen followed by maintenance treatment every 8 weeks for patients who demonstrate a clinical response (reduction in complete Mayo score of ≥ 3 points and ≥ 30% from baseline with an accompanying decrease in rectal bleeding subscore of ≥ 1 point or absolute rectal bleeding subscore of ≤ 1 point or reduction in partial Mayo score of ≥ 2 points and ≥ 25% from baseline with an accompanying decrease in rectal bleeding subscore of ≥ 1 point or absolute rectal bleeding subscore of ≤ 1 point) 6 to 8 weeks after completion of the induction regimen.
In the GEMINI I study, the induction regimen was administered at weeks 0 and 2 and maintenance dosing started at week 6. However exploratory analyses suggest a higher rate of long term clinical response and remission will be achieved with a 0, 2 and 6 week induction regimen followed by maintenance treatment every 8 weeks for patients who demonstrate a clinical response (reduction in complete Mayo score of ≥ 3 points and ≥ 30% from baseline with an accompanying decrease in rectal bleeding subscore of ≥ 1 point or absolute rectal bleeding subscore of ≤ 1 point or reduction in partial Mayo score of ≥ 2 points and ≥ 25% from baseline with an accompanying decrease in rectal bleeding subscore of ≥ 1 point or absolute rectal bleeding subscore of ≤ 1 point) 6 to 8 weeks after completion of the induction regimen.

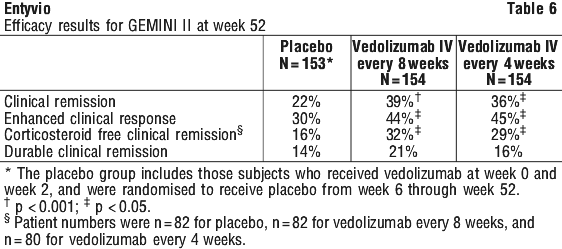 In the GEMINI II study, the induction regimen was administered intravenously at weeks 0 and 2 and maintenance dosing started at week 6. However exploratory analyses suggest a higher rate of long-term clinical response with a 0, 2 and 6 week induction regimen followed by maintenance treatment every 8 weeks for patients who demonstrate a clinical response (≥ 70-point decrease in CDAI score from baseline of induction) 6 to 8 weeks after completion of the induction regimen.
In the GEMINI II study, the induction regimen was administered intravenously at weeks 0 and 2 and maintenance dosing started at week 6. However exploratory analyses suggest a higher rate of long-term clinical response with a 0, 2 and 6 week induction regimen followed by maintenance treatment every 8 weeks for patients who demonstrate a clinical response (≥ 70-point decrease in CDAI score from baseline of induction) 6 to 8 weeks after completion of the induction regimen.

 Approximately two-thirds of patients had received prior (for UC or pouchitis) TNFα antagonist therapy (33 in vedolizumab and 31 in placebo treatment groups). Among these patients, 33.3% in the vedolizumab group achieved clinical remission at week 14 compared with 9.7% in the placebo group.
Approximately two-thirds of patients had received prior (for UC or pouchitis) TNFα antagonist therapy (33 in vedolizumab and 31 in placebo treatment groups). Among these patients, 33.3% in the vedolizumab group achieved clinical remission at week 14 compared with 9.7% in the placebo group. Do not store any unused portion of the reconstituted solution or infusion solution for reuse. Each vial is for single-use only.
Do not store any unused portion of the reconstituted solution or infusion solution for reuse. Each vial is for single-use only.