1 Name of Medicine
Eribulin mesilate.
2 Qualitative and Quantitative Composition
Each Eribulin Dr. Reddy's 1 mg/2 mL solution for injection vial contains 1 mg eribulin mesilate.
Excipient with known effect.
Alcohol 5% v/v.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Solution for injection.
Eribulin Dr. Reddy's is a clear, colourless aqueous solution for injection.
Eribulin Dr. Reddy's contains eribulin mesilate 1 mg in 2 mL (equivalent to eribulin free base 0.88 mg in 2 mL) as the active ingredient.
4.1 Therapeutic Indications
Eribulin Dr. Reddy's is indicated for the treatment of patients with locally advanced or metastatic breast cancer who have progressed after at least one chemotherapeutic regimen for advanced disease. Prior therapy should have included an anthracycline and a taxane in either the adjuvant or metastatic setting unless these are contraindicated.
Eribulin Dr. Reddy's is indicated for the treatment of patients with unresectable liposarcoma who have received prior chemotherapy for advanced or metastatic disease.
4.2 Dose and Method of Administration
Eribulin Dr. Reddy's should only be administered under the supervision of a qualified physician experienced in the appropriate use of cytotoxic medicinal products.
The recommended dose of Eribulin Dr. Reddy's as the ready to use solution is 1.4 mg/m2 which should be administered intravenously over 2 to 5 minutes on Days 1 and 8 of every 21-day cycle.
Patients may experience nausea or vomiting. Antiemetic prophylaxis including corticosteroids should be considered.
Dose delays during therapy.
The administration of Eribulin Dr. Reddy's should be delayed on Day 1 or Day 8, for a maximum of 1 week, for any of the following: absolute neutrophil count (ANC) < 1 x 109/L; platelets < 75 x 109/L; Grade 3 or 4 non-haematological toxicities.
If toxicities do not resolve or improve to ≤ grade 2 severity by Day 15, omit the dose.
If toxicities resolve or improve to ≤ grade 2 severity by Day 15, administer Eribulin Dr. Reddy's at a reduced dose (see dose reduction Table 1) and initiate the next cycle no sooner than 2 weeks later.
Dose reduction during therapy.
Dose reduction recommendations for retreatment are shown in Table 1.
 Do not re-escalate the Eribulin Dr. Reddy's dose after it has been reduced.
Do not re-escalate the Eribulin Dr. Reddy's dose after it has been reduced.
Special populations.
Hepatic impairment. Impaired liver function due to metastases.
The recommended dose of Eribulin Dr. Reddy's in patients with mild hepatic impairment (Child-Pugh A) is 1.1 mg/m2 administered intravenously over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle.
The recommended dose of Eribulin Dr. Reddy's in patients with moderate hepatic impairment (Child-Pugh B) is 0.7 mg/m2 administered intravenously over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle.
Severe hepatic impairment (Child-Pugh C) has not been studied but it is expected that a more marked dose reduction is needed if Eribulin Dr. Reddy's is used in these patients.
Impaired liver function due to cirrhosis.
This patient group has not been studied. The doses above may be used in mild and moderate impairment but close monitoring is advised as the doses may need readjustment.
Renal impairment. No dose adjustment is needed in patients with stage 2 chronic kidney disease (GFR 60-89 mL/min/1.73 m2).
In patients with moderate or severe renal impairment, reduction of the starting dose should be considered.
Physicians should exercise caution in the use of Eribulin Dr. Reddy's in patients with stage 3-5 chronic kidney disease (GFR < 60 mL/min/1.73 m2). The recommended Eribulin Dr. Reddy's dose in patients with stage 3 chronic kidney disease (GFR 30-59 mL/min/1.73 m2) or stage 4-5 chronic kidney disease (GFR < 30 mL/min/1.73 m2) is 0.7 mg/m2 administered over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle (see Section 5.2 Pharmacokinetic Properties, Renal impairment).
Elderly patients. No specific dose adjustments are recommended based on the age of the patient (see Section 4.8 Adverse Effects (Undesirable Effects)).
Paediatric patients. There is no relevant use of Eribulin Dr. Reddy's in children and adolescents in the indication of breast cancer.
The safety and efficacy of eribulin mesilate in children from birth to 18 years of age have not yet been established in soft tissue sarcoma. No data are available.
Preparation and method of administration.
Eribulin Dr. Reddy's is a ready to use solution and may be used undiluted, or the dose can be diluted, using aseptic technique, in up to 100 mL of sodium chloride 9 mg/mL (0.9%) solution for injection. It must not be mixed with other medicinal products and should not be diluted in glucose 5% infusion solution. Good peripheral venous access, or a patent central line, should be ensured prior to administration. There is no evidence that Eribulin Dr. Reddy's is a vesicant or an irritant. In the event of extravasation, treatment should be symptomatic. See Section 6.6 Special Precautions for Disposal for further details relating to handling this cytotoxic product and spill management.
During Eribulin Dr. Reddy's administration via infusion, a substantial part of the solution may be lost as a result of gravity flow therefore flush the intravenous line to ensure administration of the complete dose.
The product is for single use in one patient only. Discard any unused residue. The product is sterile and does not contain any antimicrobial preservative.
The product and diluted solution are practically free from visible particles. Discard the product if there is discolouration or visible particles.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients (see Section 6.1 List of Excipients).
Breast feeding.
4.4 Special Warnings and Precautions for Use
Haematology.
Myelosuppression is dose dependent and primarily manifested as neutropenia. In the EMBRACE study, neutropenia occurred in 82% of breast cancer patients treated with eribulin mesilate, with severe neutropenia (> Grade 3) in 57% of patients leading to discontinuation in < 1% of patients (see Section 4.8 Adverse Effects (Undesirable Effects)). In the patients treated with eribulin mesilate in soft tissue sarcoma studies (STS population), neutropenia (adverse events and laboratory abnormalities) occurred in 76.0%, with severe neutropenia (≥ Grade 3) in 53% of patients.
Monitoring of complete blood counts should be performed on all patients prior to each dose of Eribulin Dr. Reddy's. Treatment with Eribulin Dr. Reddy's should only be initiated in patients with ANC values ≥ 1.5 x 109/L and platelets > 100 x 109/L.
Febrile neutropenia occurred in < 5% of patients treated with eribulin mesilate. Patients experiencing febrile neutropenia, severe neutropenia or thrombocytopenia, should be treated according to the recommended doses (see Section 4.2 Dose and Method of Administration).
Patients with ALT or AST > 3 x ULN experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia. Although data are limited, patients with bilirubin > 1.5 x ULN also have a higher incidence of Grade 4 neutropenia and febrile neutropenia.
During clinical studies, a higher proportion of Asian and Pacific Islander subjects reported events of neutropenia and/or shifts in laboratory values for neutrophil count decreased compared with White subjects (see Section 4.8 Adverse Effects (Undesirable Effects)).
Severe neutropenia may be managed by the use of G-CSF or equivalent at the physician's discretion in accordance with relevant guidelines.
Peripheral neuropathy.
In the EMBRACE study, peripheral neuropathy occurred in 35% of breast cancer patients treated with eribulin mesilate. Severe peripheral neuropathy (> Grade 3) occurred in 8% of patients, leading to discontinuation in 5% of patients (see Section 4.8 Adverse Effects (Undesirable Effects)).
Patients should be closely monitored prior to each dose for signs of peripheral motor and sensory neuropathy. The development of severe peripheral neurotoxicity requires a delay or reduction of dose (see Section 4.2 Dose and Method of Administration).
In clinical trials, patients with pre-existing neuropathy greater than Grade 2 were excluded.
QT prolongation.
In an uncontrolled open-label ECG study in 26 patients, QT prolongation was observed on Day 8, independent of eribulin concentration, with no QT prolongation observed on Day 1. ECG monitoring is recommended if therapy is initiated in patients with congestive heart failure, bradyarrhythmias or concomitant treatment with medicinal products known to prolong the QT interval, including Class Ia and III antiarrhythmics, and electrolyte abnormalities. Hypokalaemia or hypomagnesemia should be corrected prior to initiating Eribulin Dr. Reddy's and these electrolytes should be monitored periodically during therapy. Eribulin Dr. Reddy's should be avoided in patients with congenital long QT syndrome.
Use in combination with anti-HER2 therapy.
The efficacy and safety of using eribulin mesilate in combination with anti-HER2 therapy have not been established.
Excipients.
This medicinal product contains small amounts of ethanol (alcohol), 0.1 mL per vial.
Use in hepatic impairment.
See Section 4.2 Dose and Method of Administration, Dosage adjustment in hepatic impairment; Section 4.8 Adverse Effects (Undesirable Effects), Special populations, Hepatic impairment; Section 5.2 Pharmacokinetic Properties, Special populations, Hepatic impairment.
Use in renal impairment.
See Section 4.2 Dose and Method of Administration, Dosage adjustment in renal impairment; Section 4.8 Adverse Effects (Undesirable Effects), Special populations, Renal impairment; Section 5.2 Pharmacokinetic Properties, Special populations, Renal impairment.
Use in the elderly.
The safety profile of eribulin mesilate in elderly patients (> 65 years of age) was similar to that of patients ≤ 65 years of age except for asthenia/fatigue which showed an increasing trend with age. See Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects), Special populations, Elderly.
Paediatric use.
Clinical data are not yet available in this population.
There is no relevant use of Eribulin Dr. Reddy's in children and adolescents in the indication of breast cancer.
The safety and efficacy of eribulin mesilate in children from birth to 18 years of age have not yet been established in soft tissue sarcoma. No data are available.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of other medicinal products on eribulin.
No drug-drug interactions are expected with CYP3A4 inhibitors, CYP3A4 inducers or P-glycoprotein (Pgp) inhibitors. Clinically meaningful differences in exposure (AUC) were not observed in patients with advanced solid tumours when eribulin mesilate was administered with or without ketoconazole (a strong inhibitor of CYP3A4 and a Pgp inhibitor) and when eribulin mesilate was administered with or without rifampicin (a CYP3A4 inducer).
Eribulin is a substrate of Pgp but not a substrate of BCRP, BSEP, MRP2 or MATE1 in vitro. Eribulin is mainly (up to 70%) eliminated through biliary excretion. The transport protein involved in this process is unknown (see Section 5.2 Pharmacokinetic Properties, Excretion). Based on a study of coadministration of eribulin mesilate and ketoconazole (a Pgp inhibitor), eribulin exposure is unlikely to be increased with substances inhibiting Pgp. However, coadministration of eribulin mesilate with substances inhibiting other hepatic transport proteins (e.g. organic anion transporting proteins, multidrug resistance proteins) may increase eribulin exposure. Such coadministration is not recommended at the present time.
Effects of eribulin on other drugs.
Eribulin is a CYP3A4 inhibitor in vitro, with IC50/Ki values ranging from 2 to 50 microM depending on the substrate. No in vivo data are available. Caution and monitoring for adverse events is recommended when Eribulin Dr. Reddy's is used with substances that have a narrow therapeutic window and that are eliminated mainly via CYP3A4-mediated metabolism.
Eribulin does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP2E1 enzymes or induce CYP1A2, CYP2B6, CYP2C9, CYP2C19 or CYP3A4 enzymes at relevant clinical concentrations. Eribulin is not expected to alter the plasma concentrations of drugs that are substrates of these enzymes. Eribulin down-regulated CYP2B6 and CYP1A2 expression in human hepatocytes in vitro, and the clinical relevance of this effect is unclear.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
A fertility study was not conducted with eribulin mesilate, but based on non-clinical findings in repeated dose studies where testicular toxicity was observed in both rats (hypocellularity of seminiferous epithelium with hypospermia/aspermia) and dogs (testicular hypocellularity and epididymal hypospermia/aspermia) at less than one third of the expected clinical exposure (based on AUC data), male fertility may be compromised by treatment with eribulin mesilate.
Male patients should seek advice on conservation of sperm prior to treatment because of the possibility of irreversible infertility due to therapy with Eribulin Dr. Reddy's.
(Category D)
There is no information on the use of eribulin mesilate in pregnant women. Eribulin is embryotoxic, foetotoxic, and teratogenic in rats at less than the recommended human dose (based on body surface area, mg/m2). Eribulin Dr. Reddy's should not be used during pregnancy.
In a developmental and reproductive toxicity study, pregnant rats received IV bolus injection of eribulin mesilate during organogenesis on gestation days 8, 10 and 12. Severe external or soft tissue malformations in offspring (absence of lower jaw, tongue, stomach and spleen) were observed at 0.15 mg/kg (0.64 times the recommended human dose based on body surface area, mg/m2) and increased embryofoetal death/early resorptions and decreased foetal weights were recorded at ≥ 0.1 mg/kg (≥ 0.43 times the recommended human dose based on body surface area). Maternal toxicity was observed at ≥ 0.43 times the recommended human dose based on body surface area, and included enlarged spleen, reduced body weight gain and decreased food consumption.
Women of childbearing potential.
Women of childbearing age must be advised to avoid becoming pregnant whilst they or their male partner are receiving Eribulin Dr. Reddy's and should use effective contraception during and up to 3 months after treatment.
There is no information on the excretion of eribulin or its metabolites in human or animal breast milk. A risk to newborns or infants cannot be excluded and therefore Eribulin Dr. Reddy's must not be used during breastfeeding (see Section 4.3 Contraindications).4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Summary of safety profile.
The most commonly reported undesirable effects related to eribulin mesilate are bone marrow suppression manifested as neutropenia, leukopenia, anaemia, thrombocytopenia with associated infections. New onset or worsening of pre-existing peripheral neuropathy has also been reported. Gastrointestinal toxicities, manifested as anorexia, nausea, vomiting, diarrhoea, constipation, and stomatitis are among reported undesirable effects. Other undesirable effects include fatigue, alopecia, increased liver enzymes, sepsis and musculoskeletal pain syndrome.
Clinical trials.
Breast cancer.
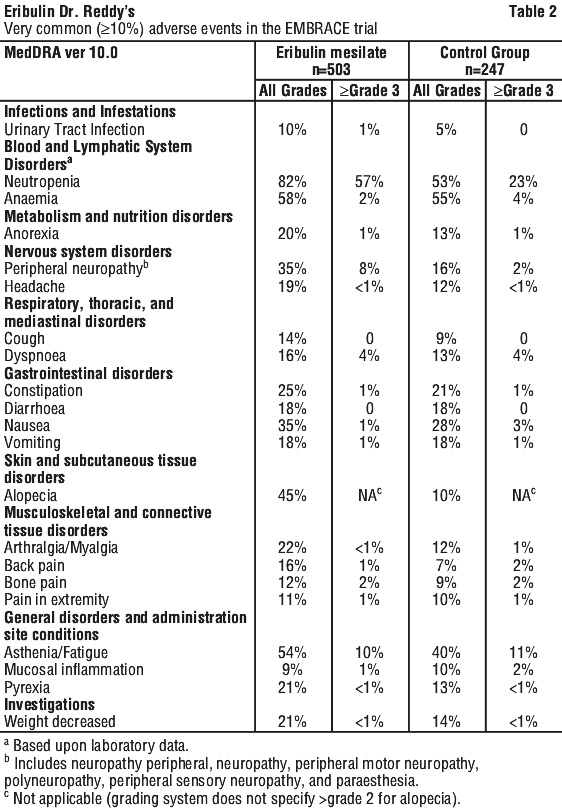
In the EMBRACE study (Study 305), 750 patients were randomised (2:1) to receive either eribulin mesilate (1.4 mg/m2 on Days 1 and 8 of a 21-day cycle) or single agent treatment chosen by their physician (control group). A total of 503 patients received eribulin mesilate, and 247 patients in the control group received therapy consisting of chemotherapy [total 97% (anthracyclines 9%, capecitabine 18%, gemcitabine 18%, taxanes 16%, vinorelbine 26%, other chemotherapies 10%)] or hormonal therapy (3%). The median duration of exposure was 118 days for patients receiving eribulin mesilate and 63 days for patients receiving control therapy. Table 2 reports the most common adverse events occurring in at least 10% of patients in either group.
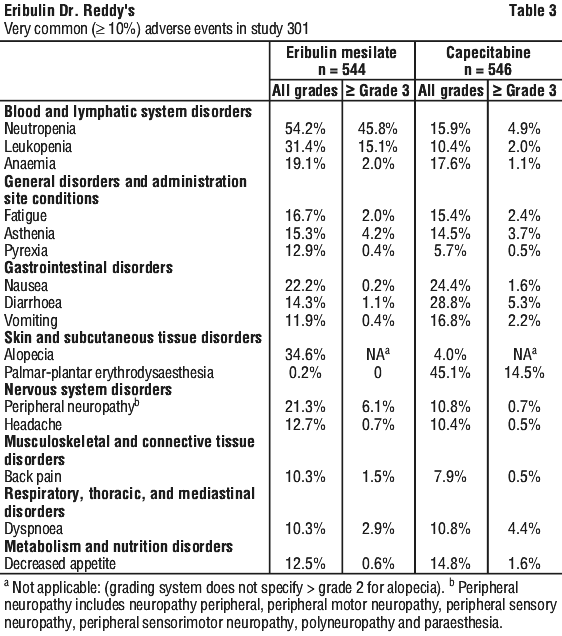
 In Study 301, 1102 patients were randomised (1:1) to receive either eribulin mesilate (1.4 mg/m2 on Days 1 and 8 of a 21-day cycle) or capecitabine (1.25 g/m2 twice daily from days 1-14 of a 21 day cycle). 12 patients in the study did not receive study treatment, so a total of 544 patients received eribulin mesilate, and 546 patients received capecitabine. The median duration of exposure was 125 days for patients receiving eribulin mesilate and 119 days for patients receiving capecitabine. Table 3 reports the most common adverse events occurring in at least 10% of patients in either group.
In Study 301, 1102 patients were randomised (1:1) to receive either eribulin mesilate (1.4 mg/m2 on Days 1 and 8 of a 21-day cycle) or capecitabine (1.25 g/m2 twice daily from days 1-14 of a 21 day cycle). 12 patients in the study did not receive study treatment, so a total of 544 patients received eribulin mesilate, and 546 patients received capecitabine. The median duration of exposure was 125 days for patients receiving eribulin mesilate and 119 days for patients receiving capecitabine. Table 3 reports the most common adverse events occurring in at least 10% of patients in either group.
Soft tissue sarcoma.
In Study 309, patients with soft tissue sarcoma were randomised (1:1) to receive either eribulin mesilate (1.4 mg/m2 on Days 1 and 8 of a 21-day cycle) or dacarbazine (850 mg/m2, 1000 mg/m2, or 1,200 mg/m2 as selected by the investigator prior to randomization according to the subject's clinical status). A total of 226 patients received eribulin mesilate, and 224 patients received dacarbazine. The median duration of exposure was 10.0 weeks for patients receiving eribulin mesilate and 9.0 weeks for patients receiving dacarbazine. Table 4 reports the most common adverse events occurring in at least 10% of patients in either group.
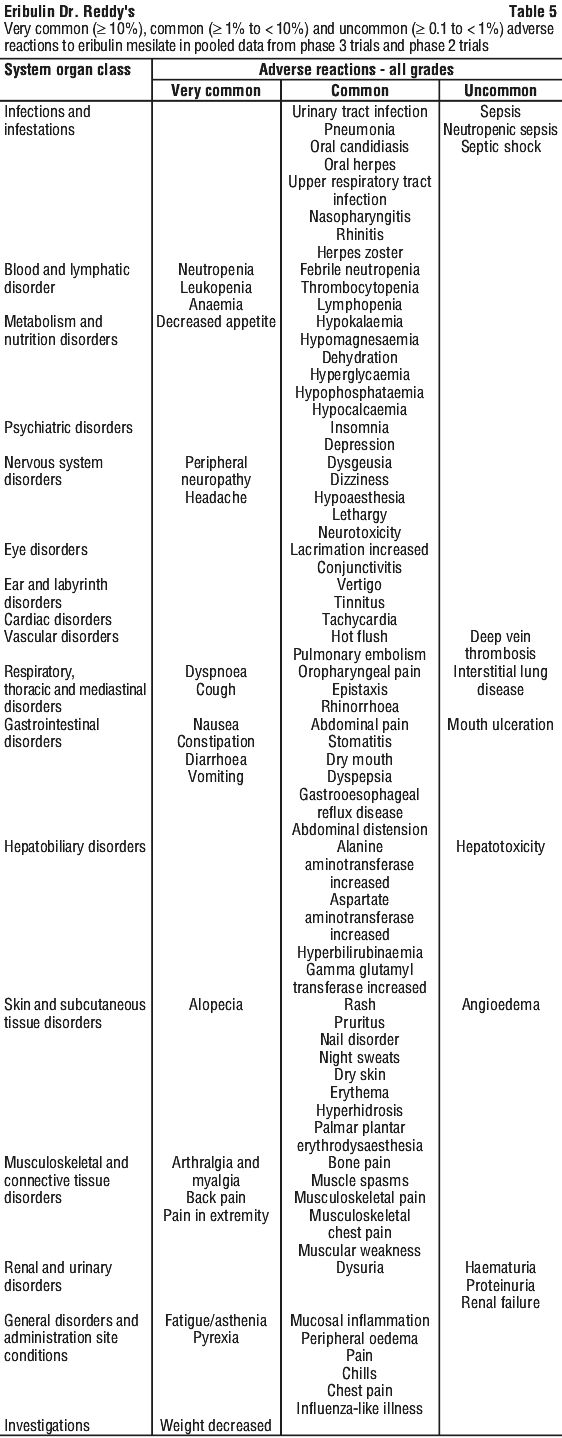
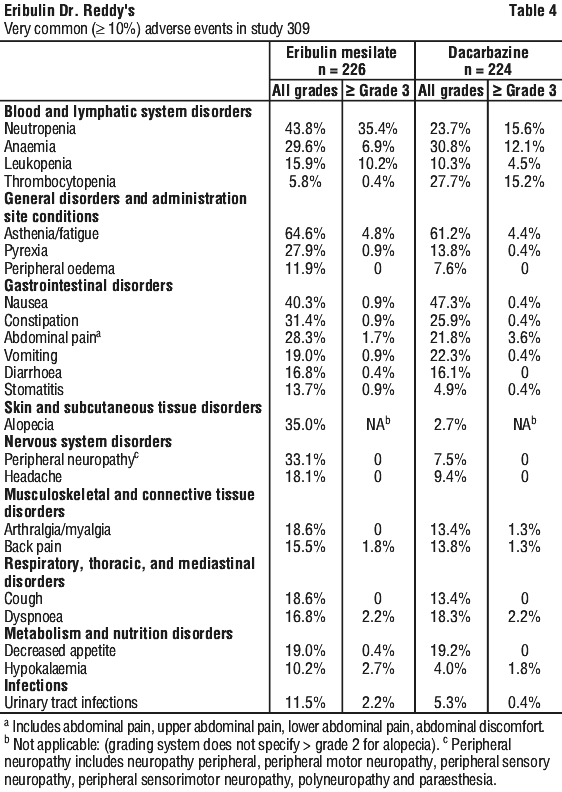 Table 5 provides a listing of very common (≥ 10%), common (≥ 1% to < 10%) and uncommon (≥ 0.1% to < 1%) adverse reactions observed in breast cancer and soft tissue sarcoma patients who received the recommended dose in Phase 2 and Phase 3 studies.
Table 5 provides a listing of very common (≥ 10%), common (≥ 1% to < 10%) and uncommon (≥ 0.1% to < 1%) adverse reactions observed in breast cancer and soft tissue sarcoma patients who received the recommended dose in Phase 2 and Phase 3 studies.
Post-marketing adverse drug reactions.
Spontaneously reported adverse effects, presented in Table 6, are reported voluntarily and it is not always possible to reliably establish frequency or a causal relationship to drug exposure.
 Overall, the safety profiles in the breast cancer and soft tissue sarcoma patient populations were similar.
Overall, the safety profiles in the breast cancer and soft tissue sarcoma patient populations were similar.
Selected adverse reactions.
Neutropenia.
The neutropenia observed was reversible and not cumulative; the mean time to nadir was 13 days and the mean time to recovery from severe neutropenia (< 0.5 x 109/L) was 8 days.
Neutrophil counts of < 0.5 x 109/L that lasted for more than 7 days occurred in 13% of breast cancer patients treated with eribulin mesilate in the EMBRACE study.
Neutropenia was reported as a Treatment Emergent Adverse Event (TEAE) in 151/404 (37.4% for all grades) in the STS population, compared with 902/1559 (57.9% for all grades) in the breast cancer population. The combined grouped TEAE and neutrophil laboratory abnormality frequencies were 307/404 (76.0%) and 1314/1559 (84.3%), respectively. The median duration of treatment was 12.0 weeks for sarcoma patients and 15.9 weeks for breast cancer patients. The incidence of Grade 3 or higher neutropenia was more common in Asian/Pacific Islander subjects (81.4%, n = 70) than in white subjects (41.0%, n = 161).
Fatal cases of febrile neutropenia, neutropenic sepsis, sepsis and septic shock have been reported. Out of 1963 breast cancer and soft tissue sarcoma patients who received eribulin mesilate at the recommended dose in clinical trials there was one fatal event each of neutropenic sepsis (0.1%) and febrile neutropenia (0.1%). In addition there were 3 fatal events of sepsis (0.2%) and one of septic shock (0.1%).
In the phase 3 EMBRACE clinical study for breast cancer, Grade 3 neutropenia occurred in 28% (143/503) of patients and Grade 4 neutropenia occurred in 29% (144/503) of patients who received eribulin mesilate. Dose reduction due to neutropenia was required in 12% (62/503) of patients. In patients with breast cancer treated with eribulin mesilate, TEAEs of Grade 3 neutropenia occurred in 23% (358/1559) of patients and Grade 4 neutropenia occurred in 27% (419/1559) of patients who received eribulin mesilate. In patients with Soft Tissue Sarcoma treated with eribulin mesilate, TEAEs of Grade 3 neutropenia occurred in 13% (51/404) of patients and Grade 4 neutropenia occurred in 19% (75/404) of patients who received eribulin mesilate.
Severe neutropenia may be managed by the use of G-CSF or equivalent at the physician's discretion in accordance with relevant guidelines.
In a phase 3 study of breast cancer patients treated with eribulin mesilate, 18% received G-CSF, compared to 26% of the eribulin mesilate treated sarcoma patients in Study 309 (see Section 4.4 Special Warnings and Precautions for Use, Haematology).
Neutropenia resulted in discontinuation in < 1% of patients receiving eribulin.
Disseminated intravascular coagulation.
Cases of disseminated intravascular coagulation have been reported, typically in association with neutropenia and/or sepsis.
Peripheral neuropathy.
In the pivotal clinical study, 17% of enrolled patients had Grade 1 peripheral neuropathy and 3% of patients had Grade 2 peripheral neuropathy at baseline. Dose reduction due to peripheral neuropathy was required by 3% (14/503) of patients who received eribulin mesilate. Four percent (20/503) of patients experienced peripheral motor neuropathy of any grade and 2% (8/503) of patients developed Grade 3 peripheral motor neuropathy.
From the pooled data on the 1559 breast cancer patients in the Phase 2 and Phase 3 studies, the most common adverse reaction resulting in discontinuation of treatment with eribulin mesilate was peripheral neuropathy (3.4%). The median time to Grade 2 peripheral neuropathy was 12.6 weeks (post 4 cycles), with the median time to Grade 2 or greater being 53 weeks. Out of the 404 sarcoma patients, 2 patients discontinued treatment with eribulin mesilate due to peripheral neuropathy. The median time to Grade 2 peripheral neuropathy was 18.4 weeks.
Development of Grade 3 or 4 peripheral neuropathy occurred in 7.4% of eribulin mesilate treated breast cancer patients and 3.5% of eribulin mesilate treated sarcoma patients. In clinical trials, patients with pre-existing neuropathy were as likely to develop new or worsening symptoms as those who entered the study without the condition.
In breast cancer patients with pre-existing Grade 1 or 2 peripheral neuropathy the frequency of treatment-emergent Grade 3 peripheral neuropathy was 14%. From the limited data available the median time to resolution of all peripheral neuropathy after the last dose was about 13 weeks (see Section 4.4 Special Warnings and Precautions for Use, Peripheral neuropathy).
Liver function test abnormalities.
Among patients with Grade 0 or 1 ALT levels at baseline, 18% of eribulin mesilate-treated patients experienced Grade 2 or greater ALT elevation. One eribulin mesilate-treated patient without documented liver metastases had concomitant Grade 2 elevations in bilirubin and ALT; these abnormalities resolved and did not recur with re-exposure to eribulin mesilate.
Special populations.
Elderly.
Of the 1559 breast cancer patients treated with the recommended dose of eribulin mesilate, 283 patients (18.2%) were ≥ 65 years of age. In the 404 sarcoma patient population, 90 patients (22.3%) treated with eribulin mesilate were ≥ 65 years of age. The safety profile of eribulin mesilate in elderly patients (> 65 years of age) was similar to that of patients ≤ 65 years of age except for asthenia/fatigue which showed an increasing trend with age. No dose adjustments are recommended for the elderly population.
Hepatic impairment.
Patients with ALT or AST > 3 x ULN experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia. Although data are limited, patients with bilirubin > 1.5 x ULN also have a higher incidence of Grade 4 neutropenia and febrile neutropenia (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.4.9 Overdose
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
In one case of overdose the patient inadvertently received 8.6 mg of eribulin mesilate (approximately 4 times the planned dose) and subsequently developed a hypersensitivity reaction (Grade 3) on Day 3 and neutropenia (Grade 3) on Day 7. Both adverse reactions resolved with supportive care.
There is no known antidote for eribulin mesilate overdose. In the event of an overdose, the patient should be closely monitored. Management of overdose should include supportive medical interventions to treat the presenting clinical manifestations.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Eribulin Dr. Reddy's (eribulin mesilate) is a first-in-class halichondrin B-based, microtubule dynamics inhibitor. It is a structurally simplified synthetic analogue of halichondrin B, a natural product isolated from the marine sponge Halichondria okadai.
Eribulin inhibits the growth phase of microtubules without affecting the shortening phase and sequesters tubulin into non-productive aggregates. Eribulin exerts its effects via a tubulin-based antimitotic mechanism leading to G2/M cell-cycle block, disruption of mitotic spindles and, ultimately, apoptotic cell death after prolonged and irreversible mitotic blockage.
Eribulin also affects the tumour microenvironment and tumour phenotype by mechanisms that are not linked to its antimitotic effects. These additional effects of eribulin include: (i) tumour vasculature remodelling whereby inner tumour cores become better perfused and less hypoxic, and (ii) phenotypic shifts from more aggressive mesenchymal phenotypes to less aggressive epithelial phenotypes via reversal of the epithelial-mesenchymal transition (EMT).
Clinical trials.
Breast cancer.
The efficacy of eribulin mesilate in breast cancer is primarily supported by two randomised Phase 3 comparative studies.
The 762 patients in the pivotal Phase 3 EMBRACE (Study 305) study had locally recurrent or metastatic breast cancer and had previously received at least two and a maximum of five chemotherapy regimens, including an anthracycline and a taxane (unless contraindicated). Patients must have progressed within 6 months of their last chemotherapeutic regimen. They were randomised 2:1 to receive either eribulin mesilate at a dose of 1.4 mg/m2 on Days 1 and 8 in a 21-day cycle administered intravenously over 2 to 5 minutes, or treatment of physician's choice (TPC), defined as any single-agent chemotherapy, hormonal treatment, or biologic therapy approved for the treatment of cancer; or palliative treatment or radiotherapy, reflecting local practice. The TPC arm consisted of 97% chemotherapy (26% vinorelbine, 18% gemcitabine, 18% capecitabine, 16% taxane, 9% anthracycline, 10% other chemotherapy), or 3% hormonal therapy. The median duration of treatment (range) for each treatment group was: vinorelbine 1.6 months (0-13.1 months); gemcitabine 2.3 months (0-14.5 months); capecitabine 3.9 months (0-21.2 months); taxane 2.9 months (0-14.5 months); anthracycline 1.9 months (0-6.5 months) and eribulin mesilate 3.9 months (0.7-16.3 months).
The primary endpoint, overall survival, was significantly better with eribulin than TPC (see Table 7). See Figure 1.
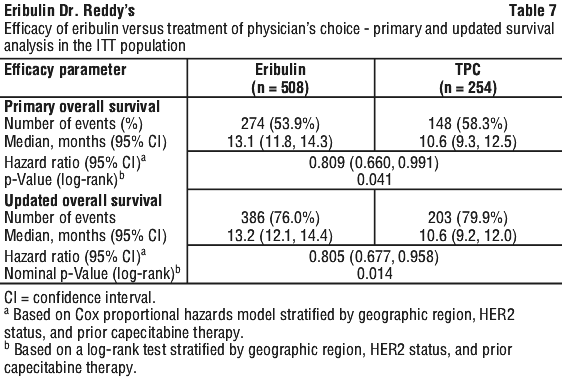
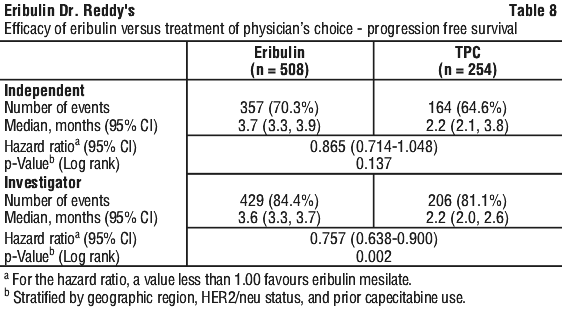
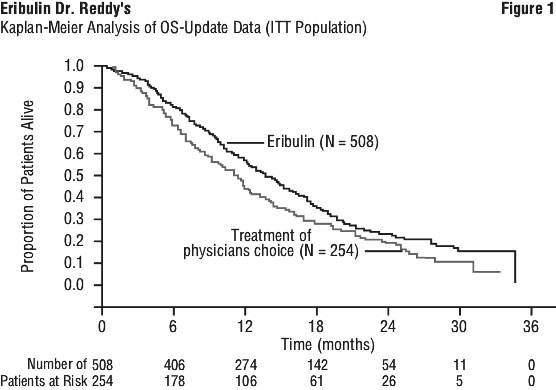 At the time of the original cut-off, analysis of progression free survival by independent and investigator review is shown in Table 8.
At the time of the original cut-off, analysis of progression free survival by independent and investigator review is shown in Table 8.
 In response evaluable patients who received eribulin mesilate, the objective response rate by the RECIST criteria was 12.2% (95% CI: 9.4%, 15.5%) by independent review and 13.2% (95% CI: 10.3%, 16.7%) by investigator review. The median response duration in this population by independent review was 4.2 months (95% CI: 3.8, 5.0 months).
In response evaluable patients who received eribulin mesilate, the objective response rate by the RECIST criteria was 12.2% (95% CI: 9.4%, 15.5%) by independent review and 13.2% (95% CI: 10.3%, 16.7%) by investigator review. The median response duration in this population by independent review was 4.2 months (95% CI: 3.8, 5.0 months).
The positive effect on OS and PFS was seen in both taxane-refractory and non-refractory groups of patients. In the OS update, the HR for eribulin mesilate versus TPC was 0.90 (95% CI 0.71, 1.14) in favour of eribulin mesilate for taxane-refractory patients and 0.73 (95% CI 0.56, 0.96) for patients not taxane-refractory. In the investigator assessment-based analysis of PFS (based on original data cut-off), the HR was 0.77 (95% CI 0.61, 0.97) for taxane-refractory patients and 0.76 (95% CI 0.58, 0.99) for patients not taxane-refractory.
The positive effect on OS was seen both in capecitabine-naïve and in capecitabine pre-treated patient groups. The analysis of updated OS showed a survival benefit for the eribulin mesilate group compared to TPC both in capecitabine pre-treated patients with a HR of 0.787 (95% CI 0.645, 0.961), and for the 11 capecitabine-naïve patients with a corresponding HR of 0.865 (95% CI 0.606, 1.233). Investigator assessment-based analysis of PFS (based on original data cut-off), also showed a positive effect in the capecitabine pre-treated group with a HR of 0.68 (0.56, 0.83). For the capecitabine-naïve group the corresponding HR was 1.03 (0.73, 1.45).
The second Phase 3 study (study 301) in earlier line metastatic breast cancer, was an open-label, randomised, study in patients (n=1102) with locally advanced or metastatic breast cancer to investigate the efficacy of eribulin monotherapy compared to capecitabine monotherapy in terms of OS and PFS as co-primary endpoint. Patients had previously received up to three prior chemotherapy regimens, including both an anthracycline and a taxane and a maximum of two for advanced disease, with the percentage who had received 0, 1 or 2 prior chemotherapy treatments for metastatic breast cancer being 20.0%, 52.0% or 27.2% respectively. The HER2 status of the patients was: 15.3% positive, 68.5% negative and 16.2% unknown, whilst 25.8% of patients were triple negative. See Figure 2.
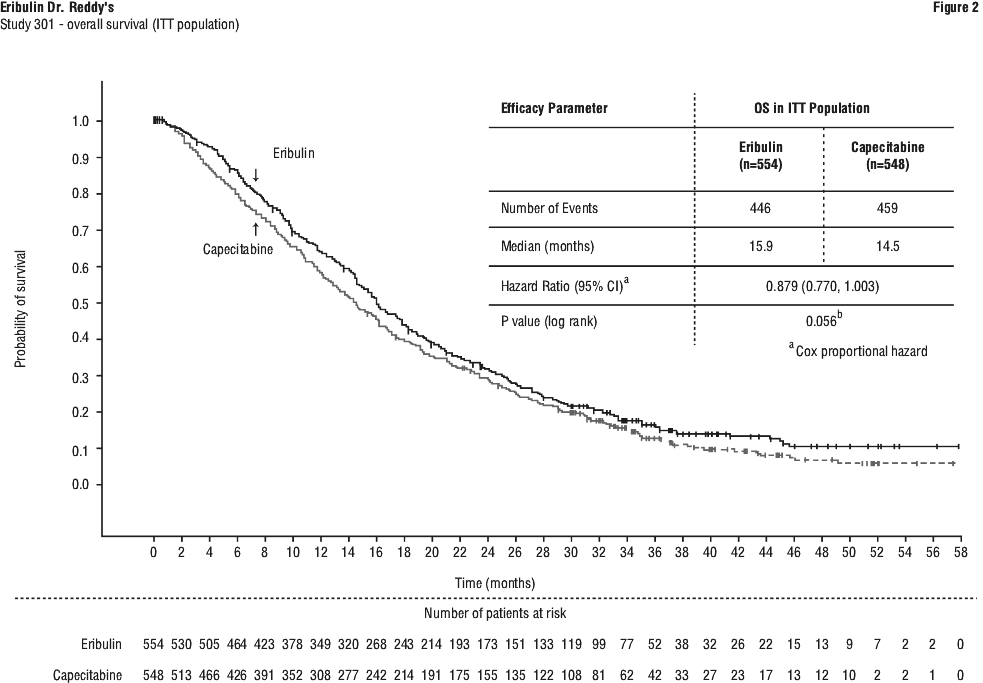 Progression free survival assessed by independent review was similar between eribulin and capecitabine with medians of 4.1 months vs 4.2 months (HR 1.08; [95% CI: 0.932, 1.250]) respectively. Objective response rate as assessed by independent review was also similar between eribulin and capecitabine; 11.0% (95% CI: 8.5, 13.9) in the eribulin group and 11.5% (95% CI: 8.9, 14.5) in the capecitabine group.
Progression free survival assessed by independent review was similar between eribulin and capecitabine with medians of 4.1 months vs 4.2 months (HR 1.08; [95% CI: 0.932, 1.250]) respectively. Objective response rate as assessed by independent review was also similar between eribulin and capecitabine; 11.0% (95% CI: 8.5, 13.9) in the eribulin group and 11.5% (95% CI: 8.9, 14.5) in the capecitabine group.
Liposarcoma.
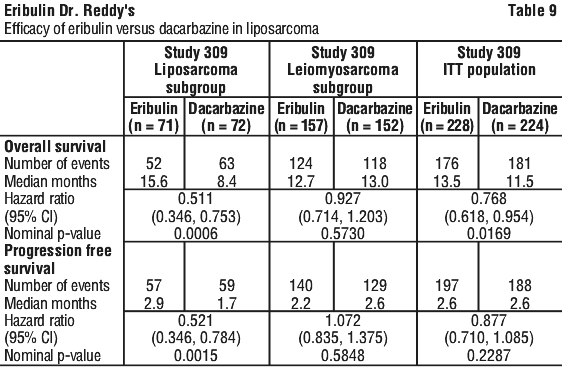
In liposarcoma the efficacy of eribulin is supported by the pivotal Phase 3 sarcoma study (Study 309). The patients (n=452) in this study had locally recurrent, inoperable and/or metastatic soft tissue sarcoma of one of two subtypes - leiomyosarcoma or liposarcoma. Patients had received at least two prior chemotherapy regimens, one of which must have been an anthracycline (unless contraindicated). Enrolment was stratified by histology, the number of prior therapies and geographic region.
Patients must have progressed within 6 months of their last chemotherapeutic regimen. They were randomised 1:1 to receive either eribulin mesilate 1.4 mg/m2 on days 1 and 8 of a 21 day cycle or dacarbazine 850 mg/m2, 1000 mg/m2 or 1200 mg/m2 (dose determined by the investigator prior to randomisation), every 21 days. Treatment continued until disease progression or unacceptable toxicity. In Study 309, a statistically significant improvement in OS was observed in patients randomised to the eribulin arm compared to the control arm. This translated into a 2 month improvement in median OS (13.5 months for eribulin mesilate treated patients vs. 11.5 months for dacarbazine treated patients). There was no significant difference in progression-free survival or overall response rate between the treatment arms in the overall population.
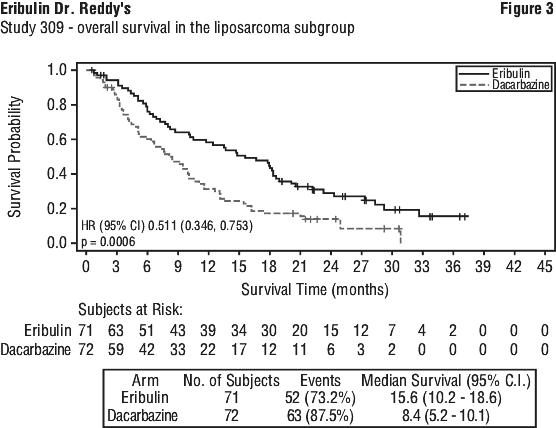
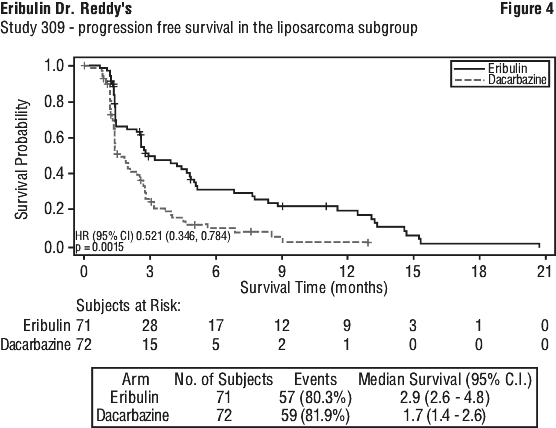
Treatment effects of eribulin mesilate were limited to patients with liposarcoma (45% dedifferentiated, 37% myxoid/round cell and 18% pleomorphic in Study 309) based on pre-planned subgroup analyses of OS and PFS. There was no difference in efficacy between eribulin and dacarbazine in patients with advanced or metastatic leiomyosarcoma. See Table 9, Figures 3-4.
Paediatric population.
No studies have been undertaken in the paediatric population in the indication of breast cancer.
No studies have been undertaken in the paediatric population in the indication of soft tissue sarcoma.
5.2 Pharmacokinetic Properties
Distribution.
The pharmacokinetics of eribulin are characterised by a rapid distribution phase followed by a prolonged elimination phase, with a mean terminal half-life of approximately 40 h. It has a large volume of distribution (range of means 43 to 114 L/m2).
Eribulin is weakly bound to plasma proteins. The plasma protein binding of eribulin (5-1000 nanogram/mL) ranged from 49% to 67% in human plasma.
Metabolism.
Unchanged eribulin was the major circulating species in plasma following administration of 14C-eribulin to patients. Metabolite concentrations represented < 0.6% of parent compound, confirming that there are no major human metabolites of eribulin.
Excretion.
Eribulin has a low clearance (range of means 1.16 to 2.42 L/h/m2). No significant accumulation of eribulin is observed on weekly administration. The pharmacokinetic properties are not dose or time dependent in the range of eribulin mesilate doses of 0.25 to 4.0 mg/m2.
Eribulin is eliminated primarily by biliary excretion. The transport protein involved in the excretion is presently unknown. Preclinical studies indicate that eribulin is transported by Pgp. However, it is unknown whether Pgp is contributing to the biliary excretion of eribulin.
After administration of 14C-eribulin to patients, approximately 82% of the dose was eliminated in faeces and 9% in urine indicating that renal clearance is not a significant route of eribulin elimination.
Unchanged eribulin represented most of the total radioactivity in faeces and urine.
Special populations.
Hepatic impairment.
A study evaluated the PK of eribulin in patients with mild (Child-Pugh A; n=7) and moderate (Child-Pugh B; n=5) hepatic impairment due to liver metastases. Compared to patients with normal hepatic function (n=6), eribulin exposure increased 1.8-fold and 2.5-fold in patients with mild and moderate hepatic impairment, respectively. Administration of eribulin mesilate at a dose of 1.1 mg/m2 to patients with mild hepatic impairment and 0.7 mg/m2 to patients with moderate hepatic impairment resulted in a somewhat higher exposure than after a dose of 1.4 mg/m2 to patients with normal hepatic function. Eribulin mesilate was not studied in patients with severe hepatic impairment (Child-Pugh C). There is no study in patients with hepatic impairment due to cirrhosis. See Section 4.2 Dose and Method of Administration for dosage recommendation.
Renal impairment.
The pharmacokinetics of eribulin were evaluated in a Phase 1 study in patients with stage 2 chronic kidney disease (GFR 60-89 mL/min/1.73 m2), stage 3 chronic kidney disease (GFR 30-59 mL/min/1.73 m2), or stage 4-5 chronic kidney disease (GFR < 30 mL/min/1.73 m2).
Increase in dose-normalised Cmax was 1.31-fold (90% CI 0.84-2.05) for stage 3 chronic kidney disease and 2.02-fold (90% CI 1.27-3.21) for stage 4-5 chronic kidney disease compared to normal renal function. Stage 3 chronic kidney disease and stage 4-5 chronic kidney disease increased mean dose normalised AUC (0-inf) 1.49-fold (90% CI 0.9-2.45) compared to normal renal function. Renal impairment therefore has an up to three fold effect on eribulin exposure and dosing modifications and extra haematology monitoring should be undertaken.
See Section 4.2 Dose and Method of Administration for treatment recommendations in renal impairment.
5.3 Preclinical Safety Data
Genotoxicity.
Eribulin was not mutagenic in the in vitro bacterial reverse mutation assay (Ames test). Eribulin was positive in the in vitro mouse lymphoma mutagenesis assay and was clastogenic in the in vivo rat micronucleus assay.
Carcinogenicity.
No carcinogenicity studies have been conducted with eribulin.6 Pharmaceutical Particulars
6.1 List of Excipients
Eribulin Dr. Reddy's solution for injection contains the excipients ethanol - absolute 0.1 mL, hydrochloric acid qs, sodium hydroxide qs and water for injections qs.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
In use storage.
If not used immediately Eribulin Dr. Reddy's as the undiluted solution in a syringe should not normally be stored longer than 4 hours at 25°C and ambient lighting, or 24 hours at 2°C-8°C. Diluted solutions of Eribulin Dr. Reddy's (0.02 mg/mL to 0.2 mg/mL in sodium chloride 9 mg/mL (0.9%)) solution for injection should not be stored longer than 24 hours at 2°C-8°C.
6.4 Special Precautions for Storage
Store below 25°C. Do not refrigerate. Do not freeze.
6.5 Nature and Contents of Container
Eribulin Dr. Reddy's solution for injection is supplied in a type I glass vial, with teflon-coated, butyl rubber stopper and flip-off aluminium over seal. The vial includes an overfill to allow withdrawal of the label claim amount. The pack sizes are cartons of 1 or 6 vials. Not all pack sizes may be marketed.
Eribulin Dr. Reddy's is a cytotoxic anticancer medicinal product and, as with other toxic compounds, caution should be exercised in its handling. The use of gloves, goggles, and protective clothing is recommended. If the skin comes into contact with the solution it should be washed immediately and thoroughly with soap and water. If it contacts mucous membranes, the membranes should be flushed thoroughly with water. Eribulin Dr. Reddy's should only be prepared and administered by personnel appropriately trained in handling of cytotoxic agents. Pregnant staff should not handle Eribulin Dr. Reddy's.
6.6 Special Precautions for Disposal
Any unused product or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Eribulin mesilate has a molecular weight of 826.0 (729.9 for free base).
Chemical structure.
The chemical name for eribulin mesilate is 11,15:18,21:24,28-triepoxy-7,9-ethano-12,15- methano-9H,15H-furo[3,2-i]furo[2',3':5,6]pyrano[4,3-b][1,4]dioxacyclopentacosin-5(4H)- one, 2-[(2S)-3-amino-2-hydroxypropyl]hexacosahydro-3-methoxy-26-methyl-20,27- bis(methylene)-,(2R,3R,3aS,7R,8aS,9S,10aR,11S,12R,13aR,13bS,15S,18S,21S,24S,26R,28R, 29aS)-, methanesulfonate (salt).
The empirical formula is C40H59NO11.CH4O3S. Eribulin mesilate has the following structural formula:

CAS number.
CAS: 441045-17-6.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription only medicine.
Summary Table of Changes
