What is in this leaflet
This leaflet answers some common questions about ERLYAND tablets. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given ERLYAND against the benefits this medicine is expected to have for you.
If you have any concerns about being given ERLYAND ask your doctor or healthcare professional.
Keep this leaflet while you are taking ERLYAND. You may need to read it again.
What ERLYAND is used for
ERLYAND is an anticancer medicine that contains the active substance apalutamide.
ERLYAND is used to treat patients with:
- prostate cancer that has not spread to other parts of the body and no longer responds to a medical or surgical treatment that lowers testosterone.
- prostate cancer that has spread to other parts of the body and still responds to treatments that lower testosterone
ERLYAND belongs to a group of medicines called androgen receptor inhibitors.
Ask your doctor or healthcare professional if you have any questions about why ERLYAND has been prescribed for you.
This medicine is available only with a doctor's prescription.
Before you take ERLYAND
When you must not use it:
Do not take ERLYAND:
- if you are allergic (hypersensitive) to apalutamide, or other ingredients of ERLYAND. See Product Description at the end of this leaflet for a list of ingredients.
Do not take ERLYAND:
- if the packaging is torn or shows signs of tampering.
- if the expiry date (month and year) printed on the pack has passed. If you take ERLYAND after the expiry date it may not work.
Do not take ERLYAND if you are pregnant or maybe potentially pregnant.
- ERLYAND is not for use in women and children.
- ERLYAND may harm your unborn baby.
Do not father a child while taking ERLYAND and for 3 months after stopping treatment.
- Use condoms and do not donate sperm during treatment and for 3 months after your treatment has finished. Talk with your healthcare provider if you have questions about birth control. If you plan to father a child, talk to your doctor or healthcare professional before taking ERLYAND.
ERLYAND should not be used by anyone under 18 years of age because it has not been studied in this age group.
Before you start to use it:
Tell your doctor if you have or have had any medical conditions, especially the following:
- have a history of seizures, brain injury, stroke, or brain tumours (non-cancerous or cancerous)
- have a history of heart disease
- you have any other medical condition
Taking other medicines:
Tell your doctor if you take any of the following medicines:
- warfarin, a medicine used to prevent blood clots
- omeprazole, a medicine to treat heartburn or gastric and duodenal ulcers
- midazolam, often used as a sedative
- rosuvastatin or gemfibrozil, medicines used to lower your lipid levels
- fexofenadine, a medicine used to treat hay fever
- rifampin, a medicine used to treat certain infections
- antifungal medicines like itraconazole or ketoconazole
ERLYAND might interact with other medicines. This may result in greater or lesser effects or even side effects from these medicines.
Tell your doctor if you are taking any other medicines, including medicines you can buy without a prescription from a pharmacy, supermarket or health food shop.
Your doctor can tell you whether you can continue the medicines you are taking or need to reduce the dose.
Taking ERLYAND
Always take ERLYAND exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
How much ERLYAND to take:
The recommended dose of ERLYAND is: four 60 mg tablets once a day.
Instructions:
- Swallow ERLYAND tablets whole with a glass of water. Do not break or chew them.
If you have trouble swallowing tablets whole:
- Stir whole ERLYAND tablets in 120 mL of applesauce. Do not crush the tablets.
- Stir after 15 minutes.
- Stir again after 30 minutes until tablets are well mixed with no chunks remaining.
- Using a spoon, swallow the mixture right away.
- Rinse the empty mixture container with 60 mL of water. Drink the water mixture and repeat the rinse with 60 mL of water one more time to make sure the medicine is swallowed completely. Drink the mixture within one hour of preparation. Do not store ERLYAND that is mixed with applesauce. - You may take them with or without food.
- Try to take ERLYAND at the same time each day.
How long to take
Take ERLYAND exactly as prescribed by your doctor or healthcare professional. Do not change your dose or stop taking ERLYAND until your doctor tells you to.
What do I do if I forget to take ERLYAND?
- If you miss a dose of ERLYAND, take your normal dose as soon as possible on the same day. Return to your normal schedule on the following day.
- Do not take extra tablets to make up the missed dose.
If you are not sure what to do, contact your doctor or pharmacist.
What do I do if I take too much? (overdose):
If you think you or anybody else has taken too much ERLYAND, contact your doctor, pharmacist or the Poisons Information Centre who will advise you what to do.
You can contact the Poisons Information Centre by dialling:
- Australia: 13 11 26
- New Zealand: 0800 POISON or 0800 764 766.
While you are taking ERLYAND
Things you must do:
Be sure to keep all your doctor's appointments so your progress can be checked.
Be sure to follow your doctor's instructions about other medicines you should take, and other things you should do.
Ask your doctor or pharmacist if you have any questions.
Tell any other doctors and pharmacists who are treating you that you are taking ERLYAND.
If you are about to be started on any new medicines, tell your doctor or pharmacist that you are taking ERLYAND.
If you have any further questions on the use of this product, ask your doctor.
Things to be careful of
Driving and using machines
There have been no studies on the effects of ERLYAND on the ability to drive or use machines however it is not anticipated that ERLYAND will affect the ability to drive and use machines.
Side Effects
Like all medicines, ERLYAND can cause side effects, although not everybody gets them. The following serious side effects may happen with this medicine:
- Seizure: treatment with ERLYAND may increase your risk of having a seizure. You should avoid activities where a sudden loss of consciousness could cause serious harm to yourself or others. Tell your healthcare provider right away if you have a loss of consciousness or seizure.
- Heart disease, stroke, or mini-stroke: blockage of the arteries in the heart or in part of the brain (which can lead to death) has happened in some people during treatment with ERLYAND. Go to the nearest emergency department right away if you get chest pain or discomfort (whether at rest or with activity) or shortness of breath, or if you get muscle weakness/paralysis in any part of the body, or difficulty in speaking during your treatment with ERLYAND.
- Fractures and falls: ERLYAND treatment can cause bones and muscles to weaken and may increase your risk of falls and fractures.
- Severe Cutaneous Adverse Reactions (SCAR): Severe rash with blisters and peeling skin, particularly around the mouth, nose, eyes, and genitals (Steven-Johnson syndrome)/Life threatening rash with blisters and peeling over much of the body (toxic epidermal necrolysis) and severe rash over the body, usually with fever and swollen lymph nodes, impacting blood cells and organs (drug reaction with eosinophilia and systemic symptoms)
- Interstitial lung disease: inflammation within the lungs that may lead to permanent damage.
- Uncomfortable feeling, with an irresistible urge to move your legs, and sometimes arms and other parts of your body.
The most common side effects include: feeling very tired, nausea, skin rash, weight loss, hot flushes, hypothyroidism, high blood pressure, decreased appetite, water retention, nausea, diarrhoea, joint pain, fractures and falls.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
If you get have any side effects, talk to your doctor or pharmacist. This includes any side effects not listed in this package insert.
Product Description
Storage
Store below 30°C. Keep tablets in the original container.
Do not store it or any other medicines in the bathroom or near a sink. Heat and dampness can destroy some medicines.
Keep this medicine out of the sight and reach of children. A locked cupboard at least one-and -a-half metres above the ground is a good place to store medicines.
Do not use this medicine after the expiry date which is stated on the package after "EXP".
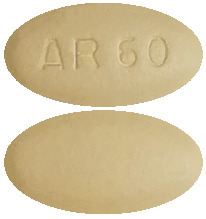
What it looks like:
The tablets are slightly yellowish to greyish green, oblong-shaped tablets with “AR 60” on one side.
ERLYAND is supplied in bottles containing 120 tablets.
Ingredients
Active ingredient:
- apalutamide
Each tablet contains 60 mg of apalutamide.
Other ingredients:
- croscarmellose sodium
- microcrystalline cellulose
- silicified microcrystalline cellulose
- colloidal anhydrous silica
- hypromellose acetate succinate
- magnesium stearate
- Opadry® II 85F210036 Green
ERLYAND tablets are gluten- and lactose- free
Sponsor
JANSSEN-CILAG Pty Ltd
1-5 Khartoum Rd
Macquarie Park
NSW 2113 Australia
Telephone: 1800 226 334
NZ Office: Auckland, New Zealand
Telephone: 0800 800 806
Registration number
60 mg tablets: AUST R 299792
This leaflet was prepared in Oct 2023.
® ERLYAND is a registered trademark of Janssen-Cilag.
Published by MIMS December 2023
 Additional clinically significant adverse reactions occurring in at least 2% of patients treated with Erlyand in the SPARTAN study included:
Additional clinically significant adverse reactions occurring in at least 2% of patients treated with Erlyand in the SPARTAN study included: White blood cell count decrease and hypertriglyceridaemia were also more common in the Erlyand arm compared to placebo in the TITAN study.
White blood cell count decrease and hypertriglyceridaemia were also more common in the Erlyand arm compared to placebo in the TITAN study. Consistent improvement in rPFS and OS was observed between subgroups based on disease volume (high vs low) and Gleason score at diagnosis (≤ 7 vs. > 7).
Consistent improvement in rPFS and OS was observed between subgroups based on disease volume (high vs low) and Gleason score at diagnosis (≤ 7 vs. > 7).


 The final analysis corroborated that treatment with Erlyand decreased the risk of symptomatic progression by 43% compared with placebo. The observed p-value (0.00000356) crossed the Obrien-Fleming (OBF) efficacy boundary (p=0.00008) for significance (see Table 4).
The final analysis corroborated that treatment with Erlyand decreased the risk of symptomatic progression by 43% compared with placebo. The observed p-value (0.00000356) crossed the Obrien-Fleming (OBF) efficacy boundary (p=0.00008) for significance (see Table 4). At the final analysis, treatment with Erlyand significantly decreased the risk of initiating cytotoxic chemotherapy by 37% compared with placebo (HR=0.629; 95% CI: 0.489, 0.808; p=0.0002) demonstrating statistically significant improvement for Erlyand versus placebo. The median time to the initiation of cytotoxic chemotherapy was not reached for either treatment arm.
At the final analysis, treatment with Erlyand significantly decreased the risk of initiating cytotoxic chemotherapy by 37% compared with placebo (HR=0.629; 95% CI: 0.489, 0.808; p=0.0002) demonstrating statistically significant improvement for Erlyand versus placebo. The median time to the initiation of cytotoxic chemotherapy was not reached for either treatment arm.
