SUMMARY CMI
EVEROLIMUS SANDOZ®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using Everolimus Sandoz?
Everolimus Sandoz contains the active ingredient everolimus. Everolimus Sandoz is used to treat various types of cancer by slowing the growth of cancer-causing cells.
For more information, see Section 1. Why am I using Everolimus Sandoz? in the full CMI.
2. What should I know before I use Everolimus Sandoz?
Do not use if you have ever had an allergic reaction to Everolimus Sandoz or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Everolimus Sandoz? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Everolimus Sandoz and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Everolimus Sandoz?
- Your doctor will tell you the dose of Everolimus Sandoz that you should take.
More instructions can be found in Section 4. How do I use Everolimus Sandoz? in the full CMI.
5. What should I know while using Everolimus Sandoz?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Everolimus Sandoz? in the full CMI.
6. Are there any side effects?
Common side effects include infections, rash, feeling weak or tired, diarrhoea, swelling of arms, hands, feet, ankles, face or other parts of the body, abdominal pain, nausea, fever, cough, headache, decreased appetite. For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
FULL CMI
EVEROLIMUS SANDOZ®
Active ingredient(s): everolimus
Consumer Medicine Information (CMI)
This leaflet provides important information about using Everolimus Sandoz. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Everolimus Sandoz.
Where to find information in this leaflet:
1. Why am I using Everolimus Sandoz?
2. What should I know before I use Everolimus Sandoz?
3. What if I am taking other medicines?
4. How do I use Everolimus Sandoz?
5. What should I know while using Everolimus Sandoz?
6. Are there any side effects?
7. Product details
1. Why am I using Everolimus Sandoz?
Everolimus Sandoz contains the active ingredient everolimus. Everolimus Sandoz is classed as an inhibitor targeting mTOR (mammalian target of rapamycin). Everolimus Sandoz is used to treat various types of cancer by slowing the growth of cancer-causing cells.
Everolimus Sandoz is used in the treatment of:
- Renal cell carcinoma, a type of kidney cancer
Everolimus Sandoz stops the cancer from making new cells and cuts off the blood supply. This slows the growth and spread of the cancer. - Neuroendocrine tumours (NETs)
Everolimus Sandoz is used to control the growth of these tumours located in the stomach and intestine, lung or pancreas. - Tuberous sclerosis complex (TSC) with angiomyolipoma of the kidney not requiring immediate surgery
Everolimus Sandoz may reduce the size of the tumour that is associated with a genetic disorder called TSC. This may lower the risk of the tumour(s) causing bleeding complications and may help to preserve kidney function. - TSC with subependymal giant cell astrocytoma (sometimes called 'SEGA'), not requiring immediate surgery
Everolimus Sandoz reduces the size of brain tumours (SEGA) that are caused by a genetic disorder called tuberous sclerosis. This may stop the tumours from causing problems as they grow, such as hydrocephalus (excessive accumulation of fluid within the brain). - Hormone receptor-positive, HER2 negative advanced breast cancer in postmenopausal women
Growth of this type of breast cancer is stimulated by oestrogens which are female sex hormones. Oestrogen inhibitors such as letrozole, anastrozole and exemestane reduce the amount of oestrogen and can slow the growth of breast cancers. After failure of letrozole or anastrozole, exemestane is used in combination with Everolimus Sandoz to prevent the breast cancer cells from becoming resistant to the exemestane. It is only used in patients whose tumour has tested negative to HER2.
2. What should I know before I use Everolimus Sandoz?
Warnings
Do not use Everolimus Sandoz if:
- you are allergic to everolimus, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use this medicine
- you have an allergy to everolimus-related medicines such as Rapamune, which contains the active ingredient sirolimus
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
Check with your doctor if you:
- have or have had problems with your liver; It may be necessary to modify your dose of Everolimus Sandoz
- have or have had diabetes or high levels of blood sugar
- have previously had Hepatitis B
- have received or about to receive radiation treatment
- have lactose intolerance
- have allergies to any other medicines, foods, preservatives or dyes
- have had or about to have recent surgery, or have an unhealed wound following surgery
- have any type of infection; it is important to treat your infection before starting Everolimus Sandoz
- are scheduled to receive any vaccine
- have any other medical conditions
- take any medicines for any other condition
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Everolimus Sandoz is not recommended for use during pregnancy.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Breastfeeding is not recommended while you are taking Everolimus Sandoz and for two weeks after the last dose of Everolimus Sandoz. It is not known whether Everolimus Sandoz passes into breast milk and could affect your baby.
Contraception
To avoid becoming pregnant, you should use effective contraception while using Everolimus Sandoz and for up to 8 weeks after you stop using it.
Children and adolescents
Everolimus Sandoz can be used in children or adolescents with normal liver function for treatment of TSC with SEGA (starting at 1 year of age).
Everolimus Sandoz is not to be used in children or adolescents for the treatment of cancer, or TSC with angiomyolipoma of the kidney.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with Everolimus Sandoz and affect how it works.
- Antibiotics such as rifampicin, rifabutin, clarithromycin and erythromycin
- Antifungal medicines such as ketoconazole, voriconazole, fluconazole and itraconazole
- Medicines for high blood pressure or heart problems such as diltiazem and verapamil
- Medicines used to treat high blood pressure or other heart problems, known as ACE inhibitors
- Medicines used to treat HIV/AIDS such as ritonavir, amprenavir, fosamprenavir, efavirenz and nevirapine
- Medicines for epilepsy such as carbamazepine, phenobarbitone and phenytoin. If you are taking a medicine to control seizures, any change in the dose of your medication may require a change in your Everolimus Sandoz dose
- St John's wort, a herbal product used to treat depression and other conditions
- Drugs used to stop the body from rejecting organ transplants such as ciclosporin
- Medicines used to prevent vomiting such as aprepitant
- Midazolam, a medicine used to treat acute seizures, or used as a sedative before or during surgery or a medical procedure
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Everolimus Sandoz.
Vaccines - tell your doctor if you need to get a vaccination. Some vaccines may be less effective if given while taking Everolimus Sandoz.
4. How do I use Everolimus Sandoz?
How much to take
Your doctor will tell you how many Everolimus Sandoz tablets to take. The dose will depend on the condition you are being treated for and your individual treatment needs.
When to take Everolimus Sandoz
- Take Everolimus Sandoz once a day, at the same time each day
- It is best to take Everolimus Sandoz in the morning, either consistently with or without food
- Do not drink grapefruit juice or eat grapefruit, star fruit or Seville oranges while taking Everolimus Sandoz. These foods can stop Everolimus Sandoz from working properly.
How to take Everolimus Sandoz
Swallow the tablets whole with a glass of water. Do not chew or crush the tablets.
If you cannot swallow Everolimus Sandoz tablets whole, you can stir the tablet into a glass of water:
- Put the required number of tablets into a glass containing approximately 30 mL of water
- Gently stir until the tablets break apart (approximately 7 minutes) then drink immediately
- Rinse the glass with the same amount of water (approximately 30 mL) and drink the contents to make sure that you get the full dose of Everolimus Sandoz.
Instructions for use and handling of tablets
Caregivers are advised to avoid contact with suspensions of Everolimus Sandoz tablets. Wash hands thoroughly before and after preparing the suspension.
If you forget to use Everolimus Sandoz
Everolimus Sandoz should be taken regularly at the same time each day. If you miss your dose at the usual time, take it as soon as you remember, and then take the next tablet as usual.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you use too much Everolimus Sandoz
If you think that you have taken too much Everolimus Sandoz, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using Everolimus Sandoz?
Things you should do
- Keep all of your doctor's appointments so that your progress can be checked
- Tell your doctor if you plan to receive a vaccine
- Avoid becoming pregnant while using Everolimus Sandoz and for up to 8 weeks after you stop using it
- Tell your doctor if you are about to have surgery, have had recent surgery or if you still have an unhealed wound following surgery.
Call your doctor straight away if you:
- Experience new or worsening cough, difficulty breathing, chest pain, shortness of breath or wheezing
- Have a temperature or chills, or another sign of an infection
- Experience pain or discomfort in the mouth or have open sores in the mouth
- Experience shortness of breath, nausea, diarrhoea, skin rashes or soreness in the mouth, gums or throat
- Become pregnant while taking this medicine
Remind any doctor, dentist or pharmacist you visit that you are using Everolimus Sandoz.
Things you should not do
- Do not stop using this medicine suddenly
- Do not take Everolimus Sandoz to treat any other complaints unless your doctor tells you to
- Do not give your medicine to anyone else, even if they have the same condition as you.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Everolimus Sandoz affects you.
Looking after your medicine
Follow the instructions in the carton on how to take care of your medicine properly.
Keep the tablets in the original packet and foils until it is time to take them.
Store below 30°C in the original packaging.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Mouth, stomach, bowel problems:
| Speak to your doctor if you have any of these less serious side effects and they worry you. If these side effects become severe, tell your doctor, pharmacist or healthcare provider |
Serious side effects
| Serious side effects | What to do |
Symptoms of an allergic reaction or infection such as:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people. Some of these side effects can only be found by laboratory testing (for example, high levels of cholesterol, lipids, or sugar in the blood).
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Everolimus Sandoz contains
| Active ingredient (main ingredient) | everolimus |
| Other ingredients (inactive ingredients) |
|
| Potential allergens | Lactose |
Do not take this medicine if you are allergic to any of these ingredients.
What Everolimus Sandoz looks like
Everolimus Sandoz tablets are available in three different strengths, supplied in packs of 30 tablets:
- Everolimus Sandoz 2.5 mg tablets are white to yellowish and elongated with no score, with "LCL" on one side and "NVR" on the other (AUST R 297503)
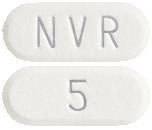
- Everolimus Sandoz 5 mg tablets are white to yellowish and elongated with no score, with "5" on one side and "NVR" on the other (AUST R 297504)
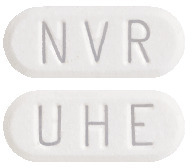
- Everolimus Sandoz 10 mg tablets are white to yellowish and elongated with no score, with "UHE" on one side and "NVR" on the other (AUST R 297505).
Who distributes Everolimus Sandoz
Everolimus Sandoz is supplied in Australia by:
Novartis Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
Macquarie Park NSW 2113
Telephone 1 800 671 203
Website: www.novartis.com.au
This leaflet was prepared in September 2021.
(eve021122c_V2.doc) based on PI (eve021122i.doc)
Published by MIMS December 2022

 For example, a patient's current dose based on BSA is 4 mg with a steady state concentration of 4 nanogram/mL. In order to achieve a target concentration above the lower Cmin limit of 5 nanogram/mL, e.g. 8 nanogram/mL, the new everolimus dose would be 8 mg (an increase of 4 mg to the current daily dose). The trough concentration should then be assessed 1 to 2 weeks after this change in dose.
For example, a patient's current dose based on BSA is 4 mg with a steady state concentration of 4 nanogram/mL. In order to achieve a target concentration above the lower Cmin limit of 5 nanogram/mL, e.g. 8 nanogram/mL, the new everolimus dose would be 8 mg (an increase of 4 mg to the current daily dose). The trough concentration should then be assessed 1 to 2 weeks after this change in dose.

 Adverse drug reaction (ADR, assessed as related to treatment by the sponsor) information is based on pooled data from 3 controlled studies but including cumulative data (N=608, including 408 patients < 18 years of age) from the double-blind plus open-label treatment phases and in addition from one non-randomised, open-label, single-arm phase II study (including open label treatment) which serve as the basis for the listed indications (see Section 4.1 Therapeutic Indications; see Table 4).
Adverse drug reaction (ADR, assessed as related to treatment by the sponsor) information is based on pooled data from 3 controlled studies but including cumulative data (N=608, including 408 patients < 18 years of age) from the double-blind plus open-label treatment phases and in addition from one non-randomised, open-label, single-arm phase II study (including open label treatment) which serve as the basis for the listed indications (see Section 4.1 Therapeutic Indications; see Table 4). The most frequent ADRs (incidence ≥ 1/10, assessed as related to treatment by the sponsor) from the pooled safety database are (in decreasing order): stomatitis, pyrexia, nasopharyngitis, diarrhoea, upper respiratory tract infection, vomiting, cough, rash, headache, amenorrhea, acne, pneumonia, urinary tract infection, sinusitis, menstruation irregular, pharyngitis, decreased appetite, fatigue, hypercholesterolemia and hypertension.
The most frequent ADRs (incidence ≥ 1/10, assessed as related to treatment by the sponsor) from the pooled safety database are (in decreasing order): stomatitis, pyrexia, nasopharyngitis, diarrhoea, upper respiratory tract infection, vomiting, cough, rash, headache, amenorrhea, acne, pneumonia, urinary tract infection, sinusitis, menstruation irregular, pharyngitis, decreased appetite, fatigue, hypercholesterolemia and hypertension.
 Overall Survival (OS) data are not mature at the time of the interim analysis and no statistically significant treatment-related difference in OS was noted [HR=0.77 (95% CI: 0.57, 1.04)].
Overall Survival (OS) data are not mature at the time of the interim analysis and no statistically significant treatment-related difference in OS was noted [HR=0.77 (95% CI: 0.57, 1.04)]. Nine-month PFS rates were 44% of patients receiving everolimus + exemestane compared with 16% in the placebo + exemestane arm at a median follow-up of 17.7 months.
Nine-month PFS rates were 44% of patients receiving everolimus + exemestane compared with 16% in the placebo + exemestane arm at a median follow-up of 17.7 months.
 Clinically or statistically significant differences were not observed between the two treatment arms in terms of time to deterioration of ECOG PS (≥ 1 point) and median times to deterioration (≥ 5%) of QLQ-C30 domain scores.
Clinically or statistically significant differences were not observed between the two treatment arms in terms of time to deterioration of ECOG PS (≥ 1 point) and median times to deterioration (≥ 5%) of QLQ-C30 domain scores.
 Eighteen-months PFS rates were 34.2% for everolimus therapy compared to 8.9% for placebo.
Eighteen-months PFS rates were 34.2% for everolimus therapy compared to 8.9% for placebo.
 In supportive analyses, positive treatment effect has been observed in all subgroups with the exception of the subgroup of patients with ileum as primary site of tumour origin (Ileum: HR=1.22 [95% CI: 0.56 to 2.65]; Non-ileum: HR=0.34 [95% CI: 0.22 to 0.54]; Lung: HR=0.43 [95% CI: 0.24 to 0.79]) (see Figure 6).
In supportive analyses, positive treatment effect has been observed in all subgroups with the exception of the subgroup of patients with ileum as primary site of tumour origin (Ileum: HR=1.22 [95% CI: 0.56 to 2.65]; Non-ileum: HR=0.34 [95% CI: 0.22 to 0.54]; Lung: HR=0.43 [95% CI: 0.24 to 0.79]) (see Figure 6). The overall response rate as per independent assessment was 2% in the everolimus arm vs. 1% in the placebo arm. Disease control rate (CR or PR or SD) for everolimus was 82.4% vs. 64.9% in the placebo arm. Tumour reduction was also evident from the corresponding waterfall plot. Results indicate that 63.6% of patients in the everolimus arm experienced tumour shrinkage versus 25.9% for placebo (Figure 7).
The overall response rate as per independent assessment was 2% in the everolimus arm vs. 1% in the placebo arm. Disease control rate (CR or PR or SD) for everolimus was 82.4% vs. 64.9% in the placebo arm. Tumour reduction was also evident from the corresponding waterfall plot. Results indicate that 63.6% of patients in the everolimus arm experienced tumour shrinkage versus 25.9% for placebo (Figure 7). The final overall survival (OS) analysis did not show statistically significant difference between those patients who received everolimus or placebo during the blinded treatment period of the study (HR = 0.90 [95% CI: 0.66 to 1.24]) (Figure 8).
The final overall survival (OS) analysis did not show statistically significant difference between those patients who received everolimus or placebo during the blinded treatment period of the study (HR = 0.90 [95% CI: 0.66 to 1.24]) (Figure 8). Clinically or statistically significant differences were not observed between the two treatment arms in terms of time to deterioration of WHO PS (HR: 1.02; 95% CI: 0.65, 1.61) and time to deterioration of FACT-G total score (HR: 0.74; 95% CI: 0.50, 1.10).
Clinically or statistically significant differences were not observed between the two treatment arms in terms of time to deterioration of WHO PS (HR: 1.02; 95% CI: 0.65, 1.61) and time to deterioration of FACT-G total score (HR: 0.74; 95% CI: 0.50, 1.10).
 Six-month PFS rates were 36% for everolimus therapy compared with 9% for placebo.
Six-month PFS rates were 36% for everolimus therapy compared with 9% for placebo. Consistent treatment effects were observed across all subgroups evaluated (i.e. EIAED use vs. EIAED non-use, sex, age, and race) at the primary efficacy analysis (Table 11).
Consistent treatment effects were observed across all subgroups evaluated (i.e. EIAED use vs. EIAED non-use, sex, age, and race) at the primary efficacy analysis (Table 11). The waterfall plots provide a graphical representation of the reduction in angiomyolipoma volume (Figure 10) at primary analysis; 95.5% of patients in the everolimus arm experienced angiomyolipoma shrinkage versus 59.4% in the placebo arm.
The waterfall plots provide a graphical representation of the reduction in angiomyolipoma volume (Figure 10) at primary analysis; 95.5% of patients in the everolimus arm experienced angiomyolipoma shrinkage versus 59.4% in the placebo arm. In the final analysis, reduction in angiomyolipoma volume improved with longer-term treatment with everolimus. At weeks 12, 96 and 192, ≥ 30% reductions in volume were observed in 75.0% (78/104), 80.6% (79/98) and 85.2% (52/61) of the treated patients, respectively. Similarly, at the same timepoints, ≥ 50% reductions in volume were observed in 44.2% (46/104), 63.3% (62/98) and 68.9% (42/61) of the treated patients, respectively.
In the final analysis, reduction in angiomyolipoma volume improved with longer-term treatment with everolimus. At weeks 12, 96 and 192, ≥ 30% reductions in volume were observed in 75.0% (78/104), 80.6% (79/98) and 85.2% (52/61) of the treated patients, respectively. Similarly, at the same timepoints, ≥ 50% reductions in volume were observed in 44.2% (46/104), 63.3% (62/98) and 68.9% (42/61) of the treated patients, respectively.
 At the primary analysis, everolimus demonstrated clinically meaningful and statistically significant improvements in skin lesion response (p=0.0002), with response rates of 26.0% (20/77) (95% CI: 16.6, 37.2) for the everolimus arm and 0% (0/37) (95% CI: 0.0, 9.5) for the placebo arm (Table 12). At the final analysis, the skin lesion response rate had increased to 68.2% (73/107) (95% CI: 58.5%, 76.9%) (Table 12), with one patient reporting a confirmed complete clinical skin lesion response and no patients experiencing progressive disease as their best response.
At the primary analysis, everolimus demonstrated clinically meaningful and statistically significant improvements in skin lesion response (p=0.0002), with response rates of 26.0% (20/77) (95% CI: 16.6, 37.2) for the everolimus arm and 0% (0/37) (95% CI: 0.0, 9.5) for the placebo arm (Table 12). At the final analysis, the skin lesion response rate had increased to 68.2% (73/107) (95% CI: 58.5%, 76.9%) (Table 12), with one patient reporting a confirmed complete clinical skin lesion response and no patients experiencing progressive disease as their best response. In an exploratory analysis of patients with TSC with angiomyolipoma who also had SEGA, the SEGA response rate (proportion of patients with ≥ 50% reduction from baseline in target lesion volumes in the absence of progression) was 10.3% (4/39) in the everolimus arm at the primary analysis (versus no responses reported in the 13 patients randomised to placebo with a SEGA lesion at baseline) and increased to 48.0% (24/50) at the final analysis.
In an exploratory analysis of patients with TSC with angiomyolipoma who also had SEGA, the SEGA response rate (proportion of patients with ≥ 50% reduction from baseline in target lesion volumes in the absence of progression) was 10.3% (4/39) in the everolimus arm at the primary analysis (versus no responses reported in the 13 patients randomised to placebo with a SEGA lesion at baseline) and increased to 48.0% (24/50) at the final analysis. Consistent treatment effects were observed across all subgroups evaluated (i.e. EIAED use vs. EIAED non-use, sex, and age) at the primary analysis (Table 14).
Consistent treatment effects were observed across all subgroups evaluated (i.e. EIAED use vs. EIAED non-use, sex, and age) at the primary analysis (Table 14). During the double-blind period, reduction of SEGA volume was evident within the initial 12 weeks of treatment with everolimus: 29.7% (22/74) of patients had ≥ 50% reductions in volume and 73.0% (54/74) of patients had ≥ 30% reductions in volume. Sustained reductions were evident at Week 24, 41.9% (31/74) of patients had ≥ 50% reductions and 78.4% (58/74) of patients had ≥ 30% reductions in SEGA volume.
During the double-blind period, reduction of SEGA volume was evident within the initial 12 weeks of treatment with everolimus: 29.7% (22/74) of patients had ≥ 50% reductions in volume and 73.0% (54/74) of patients had ≥ 30% reductions in volume. Sustained reductions were evident at Week 24, 41.9% (31/74) of patients had ≥ 50% reductions and 78.4% (58/74) of patients had ≥ 30% reductions in SEGA volume.
 Additional clinical benefits of everolimus were observed such as reductions in severity of skin lesions and size of renal angiomyolipoma.
Additional clinical benefits of everolimus were observed such as reductions in severity of skin lesions and size of renal angiomyolipoma. At the time of the primary analysis, angiomyolipoma responses were only observed in the everolimus arm (n/N:16/30; 53.3%; 95% CI: 34.3, 71.7). At the time of final analysis, among the 41 TSC-SEGA patients with an angiomyolipoma lesion(s) present at start of treatment with everolimus, 30 patients (73.2%; 95% CI: 57.1, 85.8) achieved, as their best overall response, at least a 50% reduction in sum of angiomyolipoma volumes. Among the 37 patients with evaluable angiomyolipoma tumour assessments, 35 patients (94.6%) experienced a reduction in the sum of target angiomyolipoma volumes relative to baseline as their best percentage change. Over the entire duration of the study, no new angiomyolipoma lesions were observed, nor were instances of grade 2 or worse bleeding episodes reported.
At the time of the primary analysis, angiomyolipoma responses were only observed in the everolimus arm (n/N:16/30; 53.3%; 95% CI: 34.3, 71.7). At the time of final analysis, among the 41 TSC-SEGA patients with an angiomyolipoma lesion(s) present at start of treatment with everolimus, 30 patients (73.2%; 95% CI: 57.1, 85.8) achieved, as their best overall response, at least a 50% reduction in sum of angiomyolipoma volumes. Among the 37 patients with evaluable angiomyolipoma tumour assessments, 35 patients (94.6%) experienced a reduction in the sum of target angiomyolipoma volumes relative to baseline as their best percentage change. Over the entire duration of the study, no new angiomyolipoma lesions were observed, nor were instances of grade 2 or worse bleeding episodes reported. Long-term follow-up to a median duration of 67.8 months (range: 4.7 to 83.2 months) demonstrated sustained efficacy with a median reduction in primary SEGA volume per independent central review of 0.50 cm3 at Month 60 (range: -0.74 to 9.84 cm3; n=23).
Long-term follow-up to a median duration of 67.8 months (range: 4.7 to 83.2 months) demonstrated sustained efficacy with a median reduction in primary SEGA volume per independent central review of 0.50 cm3 at Month 60 (range: -0.74 to 9.84 cm3; n=23).
