1. Why am I using Evrysdi?
Evrysdi contains the active ingredient risdiplam. Risdiplam is a selective survival of motor neuron 2 (SMN2) gene splicing modifier.
Evrysdi is used to treat spinal muscular atrophy (SMA) in patients 2 months and older.
SMA is a genetic condition people can be born with, that is caused by a shortage of a protein called “survival of motor neuron” (SMN) in the body. The SMN protein is needed for nerves to function properly. Not having enough SMN protein results in the loss of motor neurons, leading to muscle weakness and muscle wasting. Basic activities such as head and neck control, sitting, crawling and walking may be affected. The muscles used for breathing and swallowing may also be affected.
Evrysdi works by helping the body make more SMN protein and maintain SMN protein levels throughout the body with continued use. This reduces the loss of nerve cells and may improve muscle strength and function in a broad range of ages and SMA types.
In infants with SMA, Evrysdi improves survival, preserves swallowing, improves likelihood of achieving motor milestones, and reduces the need for hospitalisation and ventilator supported breathing. In children (toddlers to adolescents) and adults, Evrysdi may maintain or improve motor function over time.
2. What should I know before I use Evrysdi?
Warnings
Do not take Evrysdi if:
- you are allergic to risdiplam, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- have any other medical conditions
- have a kidney or liver disorder
- take any medicines for any other condition
- are female and pregnant or plan to become pregnant, or are breastfeeding or plan to breastfeed (see Pregnancy)
- are male and planning to donate sperm or have children (see Male fertility)
- have any issues with the structure of your eye
- have been diagnosed with Type 0 or IV SMA
- are older than 60 years or if your baby is younger than 2 months
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Contraception
- For women: You should use highly effective contraception during treatment and for 1 month after you stop treatment with Evrysdi.
- For men: If your female partner is of childbearing potential, you and your partner should both use highly effective contraception during treatment and for 4 months after treatment with Evrysdi has finished.
Pregnancy
- Evrysdi could harm your unborn baby. If you are pregnant, think you may be pregnant or are planning to have a baby, ask your doctor for advice before taking this medicine. Your doctor will consider the benefit of you taking Evrysdi against the risk to your baby.
- Before you start treatment with Evrysdi, your doctor may do a pregnancy test to confirm you are not pregnant.
- If you do become pregnant during treatment with Evrysdi, tell your healthcare provider doctor straight away. You and your doctor will decide what is best for you and your unborn baby.
Breastfeeding
Do not breastfeed while taking this medicine. This is because Evrysdi may pass into breast milk and may harm your baby.
Discuss with your doctor if you should stop breastfeeding or if you should stop taking Evrysdi.
Male fertility
Evrysdi may affect fertility in men.
Do not donate sperm during your treatment and for 4 months after your last dose of Evrysdi.
For your family planning, ask your doctor for advice.
Published by MIMS August 2021
 The physician should prescribe the appropriate pharmaceutical form according to the dose required, and the patient's needs. For patients with difficulty swallowing a whole tablet, the tablet can be dispersed in non-chlorinated drinking water (e.g. bottled water) or the powder for oral solution can be prescribed.
The physician should prescribe the appropriate pharmaceutical form according to the dose required, and the patient's needs. For patients with difficulty swallowing a whole tablet, the tablet can be dispersed in non-chlorinated drinking water (e.g. bottled water) or the powder for oral solution can be prescribed. Select the correct oral syringes (1 mL, 6 mL or 12 mL) based on the patient's dosage and remove the other oral syringes from the carton/dispensing pack.
Select the correct oral syringes (1 mL, 6 mL or 12 mL) based on the patient's dosage and remove the other oral syringes from the carton/dispensing pack.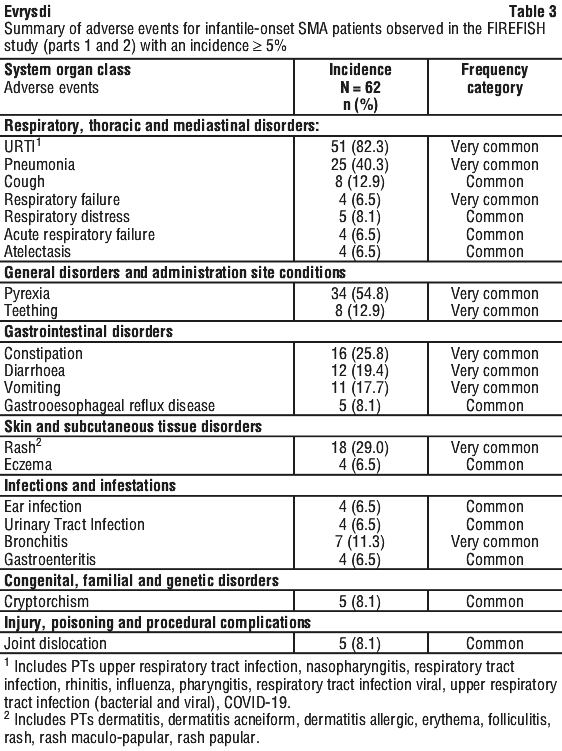

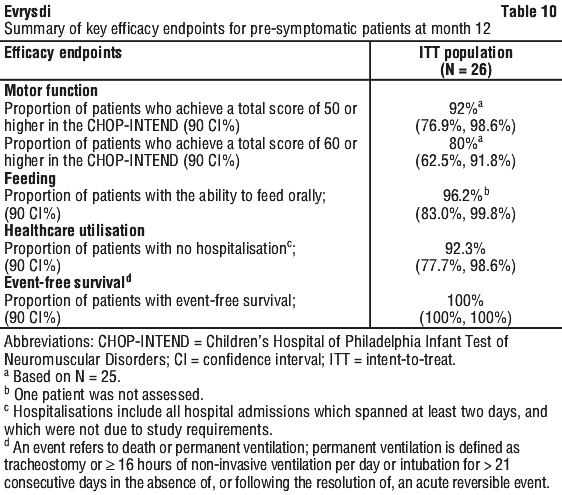
 The RAINBOWFISH study is an open-label, single-arm study that enrolled 26 patients with pre-symptomatic SMA between 16 and 41 days of age at first dose. At the primary analysis, the median exposure duration was 20.4 months (range: 10.6 to 41.9 months) (see Section 5.1, Clinical trials). The safety profile of Evrysdi in pre-symptomatic patients in the RAINBOWFISH study is consistent with the safety profile for symptomatic SMA patients treated with Evrysdi in clinical trials.
The RAINBOWFISH study is an open-label, single-arm study that enrolled 26 patients with pre-symptomatic SMA between 16 and 41 days of age at first dose. At the primary analysis, the median exposure duration was 20.4 months (range: 10.6 to 41.9 months) (see Section 5.1, Clinical trials). The safety profile of Evrysdi in pre-symptomatic patients in the RAINBOWFISH study is consistent with the safety profile for symptomatic SMA patients treated with Evrysdi in clinical trials.
 At month 24, 40% (23/58) of patients who received the therapeutic dose achieved sitting without support for 30 seconds (BSID-III, Item 26). In addition, patients continued to achieve additional motor milestones as measured by the HINE-2 at month 24; 78% of patients were able to roll (31% of patients could roll to the side, 7% could roll from prone to supine and 40% could roll from supine to prone), and 28% of patients achieved a standing measure (16% supporting weight and 12% standing with support).
At month 24, 40% (23/58) of patients who received the therapeutic dose achieved sitting without support for 30 seconds (BSID-III, Item 26). In addition, patients continued to achieve additional motor milestones as measured by the HINE-2 at month 24; 78% of patients were able to roll (31% of patients could roll to the side, 7% could roll from prone to supine and 40% could roll from supine to prone), and 28% of patients achieved a standing measure (16% supporting weight and 12% standing with support).
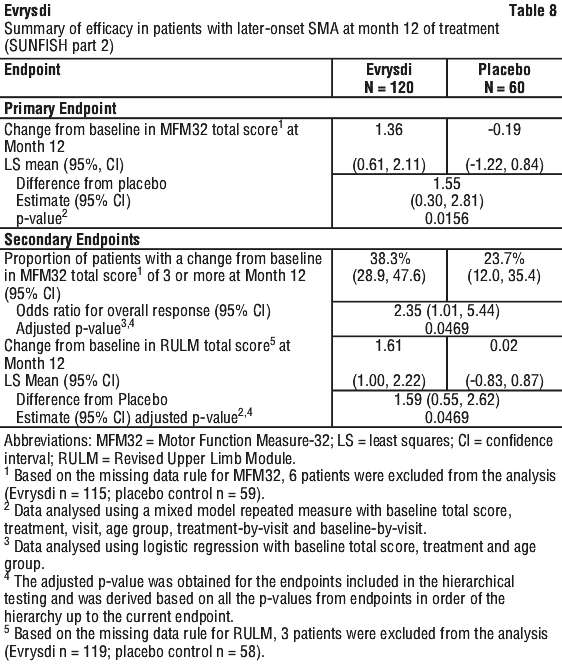 When compared to placebo, patients treated with Evrysdi demonstrated significant improvements in motor function assessed by the MFM32 after 12 months of treatment (mean difference 1.55 points; p = 0.0156). Patients 2-5 years old treated with Evrysdi demonstrated the greatest improvement on MFM32 compared to placebo control (≥ 3 points increase: 78.1% vs 52.9%). Patients ≥ 18 years old treated with Evrysdi achieved stabilisation of disease (change from baseline MFM32 total score ≥ 0 point(s): 57.1% vs. 37.5%). Consistent improvement compared to baseline MFM32 was observed in both Type 2 and 3 SMA patients treated with Evrysdi compared to placebo control (1.54 points [95% CI: 0.06, 3.02]; 1.49 points [95% CI: -0.94, 3.93], respectively).
When compared to placebo, patients treated with Evrysdi demonstrated significant improvements in motor function assessed by the MFM32 after 12 months of treatment (mean difference 1.55 points; p = 0.0156). Patients 2-5 years old treated with Evrysdi demonstrated the greatest improvement on MFM32 compared to placebo control (≥ 3 points increase: 78.1% vs 52.9%). Patients ≥ 18 years old treated with Evrysdi achieved stabilisation of disease (change from baseline MFM32 total score ≥ 0 point(s): 57.1% vs. 37.5%). Consistent improvement compared to baseline MFM32 was observed in both Type 2 and 3 SMA patients treated with Evrysdi compared to placebo control (1.54 points [95% CI: 0.06, 3.02]; 1.49 points [95% CI: -0.94, 3.93], respectively).
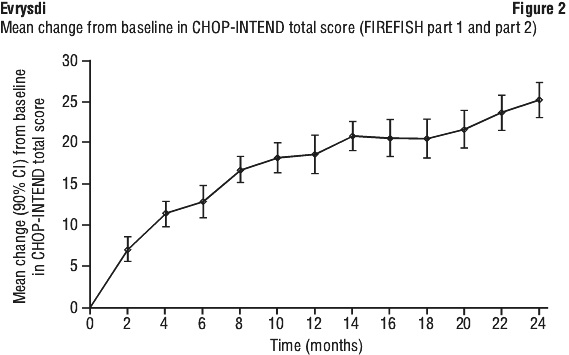
 Upon completion of 12 months of treatment, 117 patients continued to receive Evrysdi. At the time of the 24 month analysis, these patients who were treated with Evrysdi for 24 months overall experienced maintenance of improvement in motor function between month 12 and month 24. The mean change from baseline for MFM32 was 1.83 (95% CI: 0.74, 2.92) and for RULM was 2.79 (95% CI: 1.94, 3.64) at month 24. The mean change from baseline in MFM32 was -0.08 (95% CI: -1.59, 1.43) and for RULM was 2.11 (95% CI: 1.01, 3.21) at month 60.
Upon completion of 12 months of treatment, 117 patients continued to receive Evrysdi. At the time of the 24 month analysis, these patients who were treated with Evrysdi for 24 months overall experienced maintenance of improvement in motor function between month 12 and month 24. The mean change from baseline for MFM32 was 1.83 (95% CI: 0.74, 2.92) and for RULM was 2.79 (95% CI: 1.94, 3.64) at month 24. The mean change from baseline in MFM32 was -0.08 (95% CI: -1.59, 1.43) and for RULM was 2.11 (95% CI: 1.01, 3.21) at month 60. Additionally, 80% (4/5) of the primary efficacy population, 87.5% (7/8) of patients with 2 SMN2 copies, and 80.8% (21/26) of patients in the ITT population achieved sitting without support for 30 seconds (BSID-III, Item 26).
Additionally, 80% (4/5) of the primary efficacy population, 87.5% (7/8) of patients with 2 SMN2 copies, and 80.8% (21/26) of patients in the ITT population achieved sitting without support for 30 seconds (BSID-III, Item 26).