WHAT IS IN THIS LEAFLET
This leaflet answers some common questions about EYLEA. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given EYLEA against the benefits they expect it will have for you.
If you have any concerns about being given this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may need to read it again.
WHAT EYLEA IS USED FOR
EYLEA is used to treat eye conditions in adults for:
- Neovascular wet age-related macular degeneration (also known as wet AMD)
- visual impairment due to macular oedema after central retinal vein occlusion (also known as CRVO)
- visual impairment due to macular oedema after branch retinal vein occlusion (also known as BRVO)
- diabetic macular oedema (DME), which is a swelling of the retina occurring in patients with diabetes
- visual impairment due to myopic choroidal neovascularisation (also known as myopic CNV), which is associated with a severe form of short sightedness.
Wet AMD is a condition in which abnormal blood vessels grow in the back of the eye (retina). These blood vessels can leak blood and fluid into the retina and damage it leading to vision loss.
CRVO is caused by a blockage in the main blood vessel that transports blood away from the retina, in the back of your eye. The blockage stops blood from flowing in and out of the retina which causes swelling (macular oedema) and can damage your eyesight.
DME is a swelling of the retina occurring in patients with diabetes due to leakage of fluid from blood vessels within the retina. When the macula swells with fluid, central vision becomes blurry.
BRVO is caused by a blockage in one or more branches of the main blood vessel that transports blood away from the retina, in the back of your eye. The blockage stops blood from flowing in and out of the retina which causes swelling (macular oedema) and can damage your eyesight.
Myopic CNV is a severe form of myopia (short sightedness) which leads to extremely elongated eyes with additional defects in some layers in the back of the eye. This triggers the abnormal formation of new blood vessels which can cause bleeding and eventually may lead to loss of vision.
In these conditions, EYLEA can help to prevent the eyesight from becoming worse or may improve it.
Proteins called vascular endothelial growth factor-A (VEGF-A) and placental growth factor (PlGF) play an important part in the development of new blood vessels in your eye which contributes to the progression of wet AMD or myopic CNV and the development of swelling (macular oedema) due to either CRVO, BRVO or DME.
EYLEA is a type of treatment known as anti-vascular endothelial growth factor (anti-VEGF). EYLEA contains the active ingredient aflibercept, which specifically recognises and binds to VEGF-A and PlGF proteins. By blocking these proteins, EYLEA can stop the growth and leakage of abnormal blood vessels and swelling of the retina in the eye, which in turn can help improve your eyesight or stop if from getting worse.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
BEFORE YOU ARE GIVEN EYLEA
When you must not be given it
You must not be given EYLEA if you have:
- an allergy to aflibercept (the active ingredient in EYLEA) or any of the ingredients listed at the end of this leaflet. Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin. - an infection in or around your eye.
- pain or redness in your eye.
Do not give this medicine to children under the age of 18 years. There is not enough information to recommend the use of EYLEA for children or adolescents.
You must not be given this medicine after the expiry date printed on the pack. The expiry date is printed on the carton after “EXP” (for example 11 18 refers to November 2018). The expiry date refers to the last day of that month. If it has expired return it to your pharmacist for disposal.
You must not be given this medicine if the packaging is torn or shows signs of tampering. If the packaging is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should be given this medicine, talk to your doctor.
Before you are given it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you:
- think you may be allergic to aflibercept or any of the ingredients in EYLEA
- had any prior issues or problems with injections into your eyes
- have glaucoma (injection with EYLEA may trigger an increase in eye pressure in some patients within 60 minutes of the injection and your doctor may monitor this after each injection)
- have ever had a stroke or experienced transient signs of a stroke (known as a “TIA” or “mini-stroke”)
- have previously had or are planning to have an eye surgery within the past or next four weeks
If you are pregnant or plan to become pregnant or have the potential to become pregnant, please talk to your doctor. Your doctor will discuss the risks and benefits involved. It is recommended that you use effective contraception during EYLEA treatment and for at least three months after the last injection of EYLEA.
If you are breast-feeding or planning to breast-feed, EYLEA is not recommended during breast-feeding as it is not known whether EYLEA passes into breast milk.
If you have not told your doctor about any of the above, tell him/her before you are given EYLEA.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket, naturopath or health food shop. Your doctor and pharmacist have more information on medicines to be careful with or avoid when you are given this medicine.
HOW EYLEA IS GIVEN
EYLEA is given by your doctor as an injection into your eye usually under a local anaesthetic.
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions given, ask your doctor for help.
Make sure you do not wear any make up on the day of your EYLEA appointment.
On the day of your injection, your doctor or nurse will get you ready for your EYLEA treatment.
How much is given
The recommended dose of EYLEA is 50 µL (microlitre).
The interval between two doses should be no shorter than one month.
If you are being treated for wet AMD:
The injection is given once a month for the first 3 months followed by one injection every 2 months.
If considered appropriate based on your vision and test results at each visit, your doctor may decide to gradually increase or adjust the treatment interval for your next injection.
If you are being treated for impaired vision due to macular oedema caused by CRVO or BRVO:
You will start your treatment with monthly injections. After the first three injections, your doctor will determine the most appropriate treatment schedule for you based on your vision and test results at each visit. If considered appropriate, your doctor may decide to gradually increase or adjust the treatment interval for your next injection.
If you are being treated for DME:
The injection is given once a month for the first 5 months followed by one injection every 2 months.
Treatment interval may be kept at every two months or adjusted to your condition, based on your doctor’s examination. Your doctor will decide on the schedule for follow up examinations.
If you are being treated for myopic CNV:
You will start your treatment with one injection and you will receive additional injections only if, during subsequent examinations, your doctor finds that your disease persists.
How it is given
A doctor experienced in giving eye injections will inject EYLEA into your eye under sterile conditions. Before the injection your doctor will use a disinfectant eyewash to clean your eye carefully to prevent infection. Your doctor may also give you a local anaesthetic to reduce or prevent any pain you might have with the injection.
After the EYLEA injection is given, your doctor may perform some additional tests to make sure there are no complications. Eye injections like those with EYLEA can increase the pressure inside your eye.
When it is given
Your doctor will decide when you will be given EYLEA.
How long to continue treatment
Continue treatment with this medicine for as long as your doctor tells you.
Your doctor will arrange your next EYLEA appointment. Make sure you keep all of your scheduled appointments.
Your doctor may decide to stop your treatment with EYLEA if your vision is not showing benefits.
Consult your doctor before stopping treatment.
If you forget a treatment
If you miss a treatment with EYLEA, contact your doctor to make a new appointment for an examination and injection as soon as possible.
If you stop treatment with EYLEA, your vision may get worse.
If you are given too much (overdose)
If you are given more EYLEA than you need, your doctor will check the pressure in your eye and may need to treat it if it is increased.
WHILE YOU ARE GIVEN EYLEA
Things you must do
Tell your doctor if you experience any problems during the treatment, especially if you are being given EYLEA injections into both of your eyes at the same time. You may be more likely to experience side effects if you receive an injection to both of your eyes at the same time.
Tell your doctor immediately if you develop any signs or symptoms of an infection in the eye or other complications; for example, eye pain or increased discomfort, worsening eye redness, blurred or decreased vision, increased sensitivity to light. It is important to have any symptoms diagnosed and treated as soon as possible. A serious eye infection or eye disorder can sometimes develop after an injection into the eye.
Things you must not do
Do not drive or use machinery after your EYLEA injection as you may experience some temporary problems with vision.
SIDE EFFECTS
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are being treated with EYLEA.
This medicine helps most people, but it may have unwanted side effects in a few people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them. Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- visual disturbance including blurred vision or dark spots
- eye pain including injection site pain
- perception of having something floating in the eye (floaters) or sensation that something is in the eye
- increased tear production
The above list includes the more common side effects of your medicine which are usually mild and short-lived.
Tell your doctor as soon as possible if you notice any of the following:
- severe eye pain
- deterioration of eyesight
- sudden decrease in sharpness of vision in the injected eye which may be caused by tearing or detachment of the retina (the inner layer of the back of the eye)
- flashes of light and a sudden increase in floaters in the eye
- bloodshot eye caused by bleeding from small blood vessels in the outer layers of the eye or bleeding at the injection site
- hazy vision (clouding of the lens)
- damage to or swelling of the cornea (the transparent front part of the eye)
- swelling of the eyelid of the injected eye
- signs or symptoms of infection and/or inflammation of the eye
- rash, itching, hives on the skin
The above list includes serious side effects that may require medical attention.
Go to your nearest emergency room immediately if you experience any of the following:
- signs of a stroke, such as weakness or numbness of limbs or face, difficulty speaking or swallowing.
- signs of heart attack, such as chest pain which may spread to the neck and shoulders
- severe allergic reaction, such as sudden signs of rash, itching or hives on the skin; swelling of the face, lips, tongue or other parts of the body; shortness of breath, wheezing or difficulty breathing
The above are serious side effects that might need immediate medical attention or hospitalisation.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell.
Other side effects not listed above may also occur in some people. Some of these side effects (for example, an increase in the pressure inside your eye) can only be found when your doctor does tests to check your progress.
AFTER YOU ARE GIVEN EYLEA
Storage
It is unlikely you will have to store EYLEA at home.
If you do have to store it:
- Store at 2°C to 8°C. (Refrigerate. Do not freeze). Protect from light. Prior to usage, the unopened vial or pre-filled syringe blister pack may be stored at room temperature (25°C) for up to 24 hours.
- Keep the pre-filled syringe in its blister pack and carton in order to protect from light.
- Keep the vial in its carton in order to protect from light.
Do not store EYLEA or any other medicine in the bathroom, near a sink, or on a window-sill.
Do not leave it in the car.
Heat and damp can destroy some medicines.
Keep the medicine where children cannot reach it.
Disposal
Each EYLEA pre-filled syringe is to be used for one injection only and then discarded.
Each EYLEA vial and associated filter needle are to be used for one injection only and then discarded.
Return any expired or unused medicine to your pharmacist.
PRODUCT DESCRIPTION
What it looks like
EYLEA is a clear, colourless to pale yellow solution, supplied as a single dose in a glass vial or pre-filled syringe for the treatment of one eye.
Pre-filled syringe
Each carton includes a sealed blister pack with a pre-filled syringe containing an extractable volume of 90 µL (microlitre) of solution.
Vial
Each carton includes a vial containing an extractable volume of 100 µL (microlitre) of solution and a filter needle for withdrawal of the vial contents.
Not all presentations may be marketed in Australia and New Zealand.
Ingredients
Active ingredient:
- 40 mg/mL aflibercept
Inactive ingredients:
- polysorbate 20
- monobasic sodium phosphate monohydrate
- dibasic sodium phosphate heptahydrate
- sodium chloride
- sucrose
- water for injections
Sponsor
Bayer Australia Ltd
ABN 22 000 138 714
875 Pacific Highway
Pymble NSW 2073
Bayer New Zealand Ltd
PO Box 2825
Shortland Street
Auckland 1140
New Zealand
Australian registration number
EYLEA pre-filled syringe
- AUST R 180860
EYLEA vial
- AUST R 180859
Date of preparation
November 2023
See TGA website (www.ebs.tga.gov.au) in Australia or Medsafe website (www.medsafe.govt.nz) in New Zealand for latest Consumer Medicine Information.
© Bayer Australia Ltd
All rights reserved.

Published by MIMS January 2024



 The VIOLET study compared three different dosing regimens of Eylea 2 mg for treatment of DME. Following 5 consecutive monthly doses and treatment at fixed 8 week intervals for at least 1 year, patients continued treatment with Eylea 2 mg according to one of the dosing regimens:
The VIOLET study compared three different dosing regimens of Eylea 2 mg for treatment of DME. Following 5 consecutive monthly doses and treatment at fixed 8 week intervals for at least 1 year, patients continued treatment with Eylea 2 mg according to one of the dosing regimens:




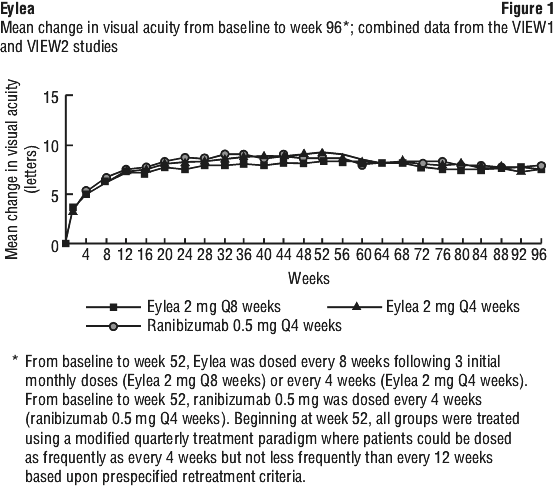 While there were small differences between Eylea and ranibizumab, no clinically relevant differences were seen between the treatment groups across all four secondary efficacy endpoints, based on the confidence intervals for the differences between Eylea and ranibizumab. All statistical tests on secondary efficacy endpoints were considered to be exploratory in the combined analysis of both studies. All secondary endpoint analyses supported the comparability of the efficacy of all 3 Eylea treatment schedules and ranibizumab.
While there were small differences between Eylea and ranibizumab, no clinically relevant differences were seen between the treatment groups across all four secondary efficacy endpoints, based on the confidence intervals for the differences between Eylea and ranibizumab. All statistical tests on secondary efficacy endpoints were considered to be exploratory in the combined analysis of both studies. All secondary endpoint analyses supported the comparability of the efficacy of all 3 Eylea treatment schedules and ranibizumab.
 No clinically meaningful differences were found between the 8Q12-, 8Q16- and 2Q8 groups in changes of NEI VFQ-25 total score at week 48 and week 96 from baseline.
No clinically meaningful differences were found between the 8Q12-, 8Q16- and 2Q8 groups in changes of NEI VFQ-25 total score at week 48 and week 96 from baseline.

 Patients in the 8Q12-, 8Q16- and 2Q8-groups who completed week 96 received a median (mean) of 9.0 (9.7), 8.0 (8.2) and 13.0 (12.8) injections, respectively.
Patients in the 8Q12-, 8Q16- and 2Q8-groups who completed week 96 received a median (mean) of 9.0 (9.7), 8.0 (8.2) and 13.0 (12.8) injections, respectively.
 At week 52, 33.3% and 33.8% of 2Q4 patients, 27.7% and 29.1% of 2Q8 patients, and 7.5% and 14.3% of laser control patients in the VIVIDDME and VISTADME studies, respectively experienced an improvement in the severity of diabetic retinopathy, as measured by a ≥ 2 step improvement in the diabetic retinopathy severity scale (DRSS). This improvement was maintained through week 100 (see Table 11).
At week 52, 33.3% and 33.8% of 2Q4 patients, 27.7% and 29.1% of 2Q8 patients, and 7.5% and 14.3% of laser control patients in the VIVIDDME and VISTADME studies, respectively experienced an improvement in the severity of diabetic retinopathy, as measured by a ≥ 2 step improvement in the diabetic retinopathy severity scale (DRSS). This improvement was maintained through week 100 (see Table 11).
 No clinically meaningful differences were found between the 8Q12-, 8Q16- and 2Q8-groups in changes of NEI VFQ-25 total score at week 48 and week 96 from baseline.
No clinically meaningful differences were found between the 8Q12-, 8Q16- and 2Q8-groups in changes of NEI VFQ-25 total score at week 48 and week 96 from baseline.
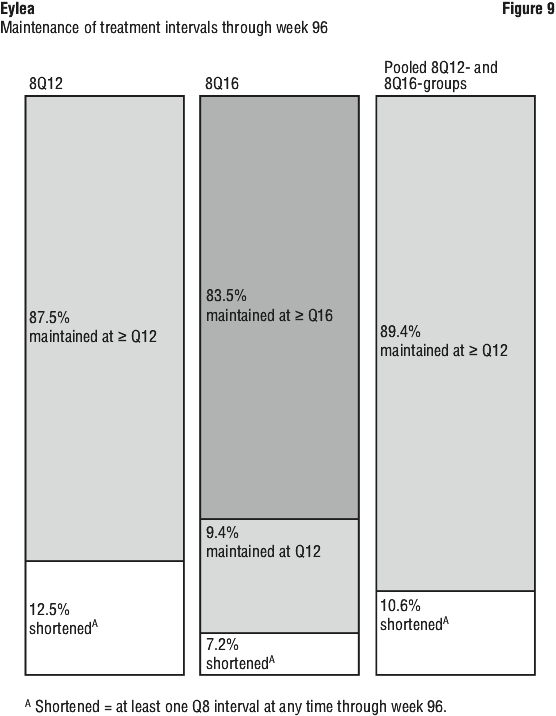
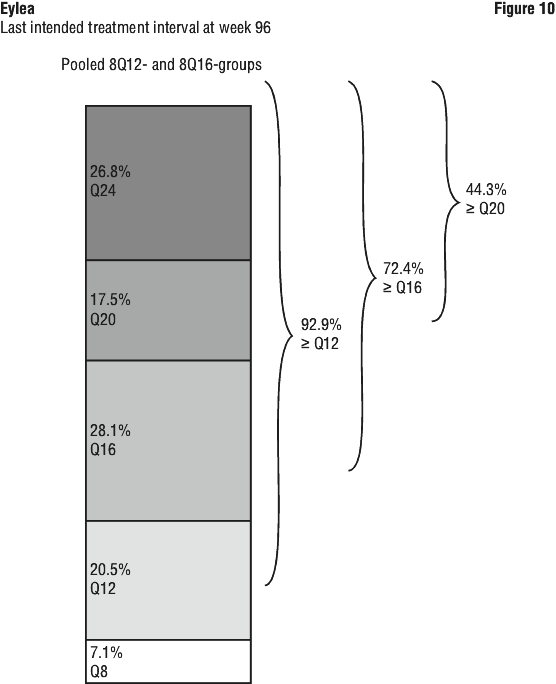 Patients in the 8Q12-, 8Q16- and 2Q8-groups who completed week 96 received a median (mean) of 9.0 (9.5), 8.0 (7.8) and 14.0 (13.8) injections, respectively.
Patients in the 8Q12-, 8Q16- and 2Q8-groups who completed week 96 received a median (mean) of 9.0 (9.5), 8.0 (7.8) and 14.0 (13.8) injections, respectively.
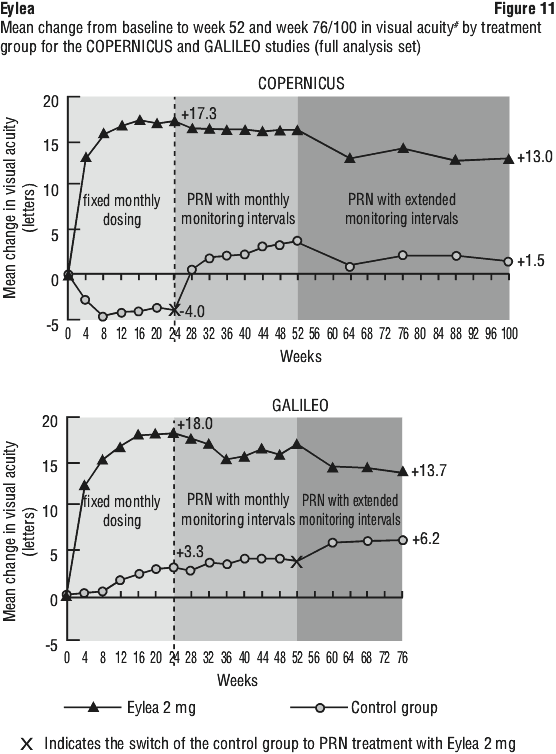 Exploratory analyses of efficacy results in all evaluable subgroups (e.g. age, gender, race, baseline visual acuity, retinal perfusion status, CRVO duration) in each study were in general consistent with the results in the overall populations.
Exploratory analyses of efficacy results in all evaluable subgroups (e.g. age, gender, race, baseline visual acuity, retinal perfusion status, CRVO duration) in each study were in general consistent with the results in the overall populations.
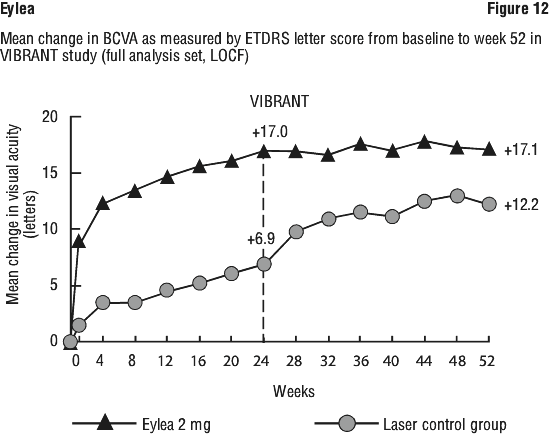 The proportion of retinal perfused patients in the Eylea group at baseline was 60.4% (n = 55). At week 24, this proportion increased to 80.2% (n = 65) and was sustained at week 52 (77.9%, n = 67). The proportion of perfused patients that started on grid laser photocoagulation was 68.9% (n = 62) at baseline. Perfusion at the week 24 primary endpoint in the laser group was 67.1% (n = 55). Patients in the laser group were eligible for rescue treatment with Eylea beginning at week 24 according to prespecified criteria. At week 52, 78.0% (n = 64) were perfused at this time.
The proportion of retinal perfused patients in the Eylea group at baseline was 60.4% (n = 55). At week 24, this proportion increased to 80.2% (n = 65) and was sustained at week 52 (77.9%, n = 67). The proportion of perfused patients that started on grid laser photocoagulation was 68.9% (n = 62) at baseline. Perfusion at the week 24 primary endpoint in the laser group was 67.1% (n = 55). Patients in the laser group were eligible for rescue treatment with Eylea beginning at week 24 according to prespecified criteria. At week 52, 78.0% (n = 64) were perfused at this time.
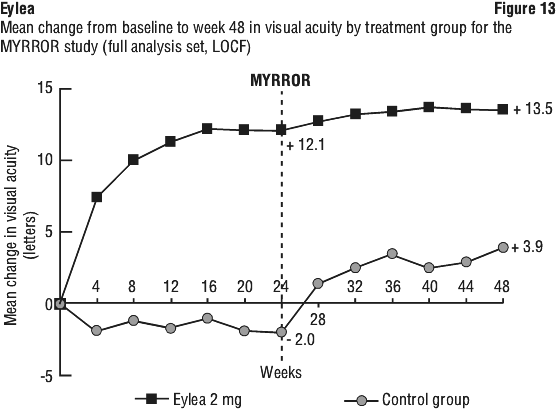 Treatment effects in all evaluable subgroups were in general, consistent with the results in the overall populations.
Treatment effects in all evaluable subgroups were in general, consistent with the results in the overall populations. The mean maximum plasma concentration of free aflibercept is approximately 50 to 500 times below the aflibercept concentration required to inhibit the biologic activity of systemic VEGF by 50% in animal models. It is estimated that after intravitreal administration of 2 mg to patients, the mean maximum plasma concentration of free aflibercept is more than 100-fold lower than the concentration of aflibercept required to half maximally bind systemic VEGF. Therefore, systemic pharmacodynamic effects are unlikely.
The mean maximum plasma concentration of free aflibercept is approximately 50 to 500 times below the aflibercept concentration required to inhibit the biologic activity of systemic VEGF by 50% in animal models. It is estimated that after intravitreal administration of 2 mg to patients, the mean maximum plasma concentration of free aflibercept is more than 100-fold lower than the concentration of aflibercept required to half maximally bind systemic VEGF. Therefore, systemic pharmacodynamic effects are unlikely.
 Chemical name: Vascular endothelial growth factor receptor type VEGFR-1 (synthetic human immunoglobulin domain 2 fragment) fusion protein with vascular endothelial growth factor receptor type VEGFR-2 (synthetic human immunoglobulin domain 3 fragment) fusion protein with immunoglobulin G1 (synthetic Fc fragment), dimer. Des-432-lysine-[human vascular endothelial growth factor receptor 1-(103-204)-peptide (containing Ig-like C2-type 2 domain) fusion protein with human vascular endothelial growth factor receptor 2-(206-308)-peptide (containing Ig-like C2-type 3 domain fragment) fusion protein with human immunoglobulin G1-(227 C-terminal residues)-peptide (Fc fragment)], (211-211':214-214')-bisdisulfide dimer.
Chemical name: Vascular endothelial growth factor receptor type VEGFR-1 (synthetic human immunoglobulin domain 2 fragment) fusion protein with vascular endothelial growth factor receptor type VEGFR-2 (synthetic human immunoglobulin domain 3 fragment) fusion protein with immunoglobulin G1 (synthetic Fc fragment), dimer. Des-432-lysine-[human vascular endothelial growth factor receptor 1-(103-204)-peptide (containing Ig-like C2-type 2 domain) fusion protein with human vascular endothelial growth factor receptor 2-(206-308)-peptide (containing Ig-like C2-type 3 domain fragment) fusion protein with human immunoglobulin G1-(227 C-terminal residues)-peptide (Fc fragment)], (211-211':214-214')-bisdisulfide dimer.