1 Name of Medicine
Iptacopan.
2 Qualitative and Quantitative Composition
Each capsule contains 200 mg iptacopan (as 225.8 mg iptacopan hydrochloride monohydrate).
List of excipients with known effect.
Gelatin may contain residual sulfites.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Capsule, hard.
Pale yellow opaque, imprinted with "LNP200" on the body and "NVR" on the cap, containing white or almost white to pale purplish-pink powder.
4.1 Therapeutic Indications
Fabhalta is indicated for the treatment of adult patients with:
paroxysmal nocturnal haemoglobinuria (PNH);
complement 3 glomerulopathy (C3G).
4.2 Dose and Method of Administration
Dosage regimen.
The recommended dose is 200 mg taken orally twice daily.
If a dose or doses are missed, the patient should be advised to take one dose of Fabhalta as soon as possible (even if it is soon before the next scheduled dose) and then to resume the regular dosing schedule.
PNH patients switching from anti-C5 (eculizumab, ravulizumab) or other PNH therapies to Fabhalta.
To reduce the potential risk of haemolysis with abrupt treatment discontinuation:
For patients switching from eculizumab, Fabhalta should be initiated no later than 1 week after the last dose of eculizumab.
For patients switching from ravulizumab, Fabhalta should be initiated no later than 6 weeks after the last dose of ravulizumab.
When switching from other PNH therapies to Fabhalta, the dosing interval and mode of action of the previous medicinal products should be considered.
Switches from complement inhibitors other than eculizumab and ravulizumab have not been studied.
Adherence to dosing schedule.
Healthcare providers should advise all patients about the importance of adherence to the dosing schedule.
In PNH patients, adherence is important to minimize the risk of haemolysis. Patients with PNH who have missed several consecutive doses should be monitored for potential signs and symptoms of haemolysis (see Section 4.4 Special Warnings and Precautions for Use). PNH is a disease that requires chronic treatment and discontinuation of this medicinal product is not recommended unless clinically indicated.
Renin angiotension system (RAS) inhibitor in C3G patients.
In the adult phase 3 study for complement 3 glomerulopathy, all patients were required to be treated with a maximally recommended or tolerated dose of angiotension converting enzyme inhibitor (ACEI) or angiotension receptor blocker (ARB) for at least 90 days prior to iptacopan initiation and throughout treatment. See Section 5.1 Pharmacodynamic Properties, Complement 3 glomerulopathy.
Special populations.
Renal impairment.
No dose adjustment is required in patients with mild (estimated glomerular filtration rate [eGFR] 60 - < 90 mL/min/1.73 m2) or moderate (eGFR 30 - < 60 mL/min/1.73 m2) renal impairment (see Section 5.2 Pharmacokinetic Properties, Special populations). No data are currently available in patients with severe renal impairment or on dialysis and no dose recommendations can be given.
Hepatic impairment.
The use of iptacopan is not recommended in patients with severe hepatic impairment (Child-Pugh class C). No dose adjustment is required for patients with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment (see Section 5.2 Pharmacokinetic Properties, Special populations).
Paediatric patients.
The safety and efficacy of Fabhalta in patients below the age of 18 years have not been established.
Geriatric patients (65 years of age or above).
Limited numbers of patients aged over 65 years were included in the clinical studies, and therefore evidence in this population is limited. No dose adjustment is required for patients aged 65 years and over (see Section 5.2 Pharmacokinetic Properties, Special populations).
Method of administration.
For oral use.
Fabhalta may be taken with or without food (see Section 5.2 Pharmacokinetic Properties).4.3 Contraindications
Fabhalta is contraindicated:
in patients with hypersensitivity to iptacopan or to any of the excipients;
in patients who are not currently vaccinated against Neisseria meningitidis and Streptococcus pneumoniae unless the risk of delaying Fabhalta treatment outweighs the risk of developing an infection from these encapsulated bacteria (see Section 4.4 Special Warnings and Precautions for Use);
for initiation in patients with unresolved serious infection caused by encapsulated bacteria, including Streptococcus pneumoniae, Neisseria meningitidis, or Haemophilus influenzae type B;
for use in combination with other complement inhibitor therapies, unless medically justified.
4.4 Special Warnings and Precautions for Use
Serious infections caused by encapsulated bacteria.
The use of complement inhibitors, such as Fabhalta, may predispose individuals to serious, life-threatening, or fatal infections caused by encapsulated bacteria. To reduce the risk of infection, all patients must be vaccinated against encapsulated bacteria, including Neisseria meningitidis, Streptococcus pneumoniae and Haemophilus influenzae type B. Refer to current local vaccination guidelines such as the Australian Immunisation Handbook.
Vaccines should be administered at least 2 weeks prior to administration of the first dose of Fabhalta. If Fabhalta must be initiated prior to vaccination, patients should be provided with antibacterial drug prophylaxis until 2 weeks after vaccine administration and vaccinated as soon as possible.
If necessary, patients may be revaccinated in accordance with current local vaccination guidelines such as the Australian Immunisation Handbook.
Vaccination reduces, but does not eliminate, the risk of serious infection. Serious infection may rapidly become life-threatening or fatal if not recognized and treated early. Patients should be informed of and monitored for early signs and symptoms of serious infection. Patients should be immediately evaluated and treated if infection is suspected.
PNH laboratory monitoring.
Patients with PNH receiving iptacopan should be monitored regularly for signs and symptoms of haemolysis, including measuring lactate dehydrogenase (LDH) levels.
Monitoring of PNH manifestations after discontinuation of Fabhalta.
If treatment with Fabhalta must be discontinued, patients should be closely monitored for signs and symptoms of haemolysis for at least 2 weeks after the last dose. These signs include elevated lactate dehydrogenase (LDH) levels along with sudden decrease in haemoglobin or PNH clone size, fatigue, haemoglobinuria, abdominal pain, dyspnoea, major adverse vascular events (including thrombosis), dysphagia, or erectile dysfunction. If discontinuation of Fabhalta is necessary, consider alternative therapy.
If haemolysis occurs after discontinuation of Fabhalta, restarting Fabhalta or initiating another treatment for PNH should be considered.
Treatment of patients with C3G.
Patients with C3G treated with immunosuppressant medicinal products may show modest proteinuria reduction with iptacopan, which is likely linked to a more treatment-resistant nature of C3G in these patients.
There is no experience with the use of iptacopan in patients with native kidney C3G who have proteinuria below 1 g/g at treatment initiation. There is limited experience with the use of iptacopan in patients with recurrent C3G after transplantation. See Section 4.8 Adverse Effects (Undesirable Effects), Complement 3 glomerulopathy; Section 5.1 Pharmacodynamic Properties, Complement 3 glomerulopathy, Clinical trials, X2202 and roll-over extension study.
Use in hepatic impairment.
See Section 5.2 Pharmacokinetic Properties, Special populations.
Use in renal impairment.
See Section 5.2 Pharmacokinetic Properties, Special populations.
Use in the elderly.
Limited numbers of patients aged over 65 years were included in the clinical studies, and therefore evidence in this population is limited. No dose adjustment is required for patients aged 65 years and over (see Section 5.2 Pharmacokinetic Properties, Special populations).
Paediatric use.
The safety and efficacy of Fabhalta in patients below the age of 18 years have not been established.
Effects on laboratory tests.
See Section 4.8 Adverse Effects (Undesirable Effects).
Co-administration with other medicinal products.
Concomitant use of iptacopan with strong inducers of CYP2C8, UGT1A1, PgP, BCRP and OATP1B1/3 has not been studied clinically; therefore, concomitant use is not recommended due to the potential for reduced efficacy of iptacopan (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). If an alternative concomitant medicinal product cannot be identified, patients with PNH should be monitored for potential signs and symptoms of haemolysis.
Educational materials.
All physicians who intend to prescribe Fabhalta must ensure they have received and are familiar with the physician educational materials. Physicians must explain and discuss the benefits and risks of Fabhalta therapy with the patient and provide them with the patient/caregiver guide and patient safety card. The patient should be instructed to seek prompt medical care if they experience any sign or symptom of serious infection or serious haemolysis (patients with PNH) following treatment discontinuation.4.5 Interactions with Other Medicines and Other Forms of Interactions
In vitro studies indicate that iptacopan does not inhibit common cytochrome P450 enzymes (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 or 3A4/5) or induce CYP1A2, 2B6, 2C9 or 3A4 at clinically relevant concentrations. Iptacopan also does not inhibit the transporters BCRP, BSEP, MATE1, MATE2-K, MRP2, OATP1B3, OAT1, OAT3, OCT1 or OCT2 at clinically relevant concentrations in vitro. Accordingly, no notable interactions are anticipated in patients. Iptacopan is a substrate for CYP2C8 and for the transporters P-glycoprotein, BCRP, MRP2, OATP1B1 and OATP1B3.
A dedicated drug interaction study in which iptacopan was co-administered with other drugs was conducted in healthy volunteers and did not demonstrate any clinically relevant interactions:
When co-administered with clopidogrel (a moderate CYP2C8 inhibitor), iptacopan Cmax and AUC increased by 5% and 36%, respectively.
When co-administered with cyclosporine (a strong OATP 1B1/1B3 inhibitor), iptacopan Cmax and AUC increased by 41% and 50%, respectively.
In the presence of iptacopan, the Cmax of digoxin (a PgP substrate) increased by 8% while its AUC was unchanged.
In the presence of iptacopan, the Cmax and AUC of rosuvastatin (an OATP substrate) remained unchanged.
Effects of other medicinal products on iptacopan.
Strong inducers of CYP2C8, UGT1A1, PgP, BCRP and OATP1B1/3.
Although concomitant administration of iptacopan with strong inducers of CYP2C8, UGT1A1, PgP, BCRP and OATP1B1/3, such as rifampicin, has not been studied clinically, concomitant use with iptacopan is not recommended due to the potential for reduced efficacy of iptacopan (see Section 4.4 Special Warnings and Precautions for Use).
Effects of iptacopan on other medicinal products.
CYP3A4 substrates.
In vitro data showed iptacopan has potential for induction of CYP3A4 and may decrease the exposure of sensitive CYP3A4 substrates. The concomitant use of iptacopan and sensitive CYP3A4 substrates has not been studied clinically. Caution should be exercised if co-administration of iptacopan with sensitive CYP3A4 substrates is required, especially for those with a narrow therapeutic index (e.g. carbamazepine, ciclosporin, ergotamine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus).
CYP2C8 substrates.
In vitro data showed iptacopan has potential for time-dependent inhibition of CYP2C8 and may increase the exposure of sensitive CYP2C8 substrates, such as repaglinide, dasabuvir or paclitaxel. The concomitant use of iptacopan and sensitive CYP2C8 substrates has not been studied clinically. Caution should be exercised if co-administration of iptacopan with sensitive CYP2C8 substrates is required.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no clinical data on the effect of Fabhalta on fertility.
Iptacopan did not impair fertility in male rats with oral administration up to the highest dose tested (750 mg/kg/day), which corresponds to 19-fold the plasma AUC for unbound drug in patients at the maximum recommended human dose (MRHD) of 200 mg twice daily. Reversible histopathological changes in the male reproductive system (testicular tubular degeneration and cell debris in the lumen of the epididymis) were observed in repeat-dose toxicity studies after oral administration in rats and dogs at doses ≥ 10-fold the MRHD based on unbound plasma AUC, with no apparent effects on sperm numbers, morphology or motility.
Iptacopan did not affect fertility in female rats with oral administration up to the highest dose tested (1000 mg/kg/day), yielding exposure 18-fold that of patients at the MRHD (based on plasma AUC for unbound drug). However, this dose did cause adverse effects on early embryonic developmental study (increased pre- and post-implantation losses and, consequently, decreased numbers of live embryos). The dose of 300 mg/kg/day is the no-observed-adverse-effect-level (NOAEL) which corresponds to ~7-fold the MRHD based on plasma AUC for unbound iptacopan.
(Category B1)
There are insufficient data on Fabhalta use in pregnant women to inform a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with untreated PNH and C3G in pregnancy. Paroxysmal nocturnal haemoglobinuria in pregnancy is associated with adverse maternal outcomes, including worsening cytopenia, thrombotic events, infections, bleeding, miscarriages and increased maternal mortality, as well as adverse fetal outcomes, including fetal death and premature delivery. Complement 3 glomerulopathy in pregnancy may be associated with adverse maternal outcomes, in particular preeclampsia and miscarriage, as well as adverse fetal outcomes including prematurity and low birth weight. The use of Fabhalta in pregnant women or women planning to become pregnant may be considered following an assessment of the risks and benefits.
No malformations of other adverse effects on embryofetal development were observed in rats or rabbits with oral administration of iptacopan during the major period of organogenesis up to the highest doses tested (1000 mg/kg/day and 450 mg/kg/day in the respective species). These doses yield exposure to iptacopan 18-times higher in rats (based on unbound plasma AUC) and 8-times higher in rabbits (based on AUC for total drug) than in patients at the MRHD.
It is not known if iptacopan and/or its metabolites are transferred into milk after oral administration in either humans or animals. There are no data on the effects of Fabhalta on the breastfed child or on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for Fabhalta and any potential adverse effects (e.g. serious infections from encapsulated bacteria) on the breastfed child from Fabhalta or from the underlying maternal condition.4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Paroxysmal nocturnal hemoglobinuria.
Summary of the safety profile.
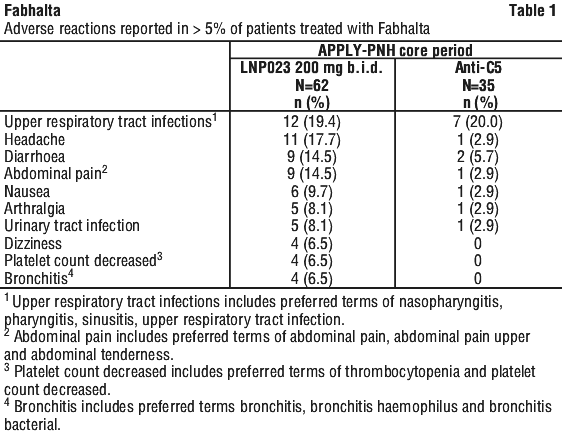
The safety profile of Fabhalta is based on analysis of safety data from 102 patients with PNH treated with Fabhalta 200 mg twice daily across two Phase 3 studies (APPLY-PNH and APPOINT-PNH). The median duration of Fabhalta exposure was 5.6 months in the core period of each study. The most commonly reported adverse reactions in patients treated with Fabhalta in APPLY-PNH (N=62) and APPOINT-PNH (N=40) were upper respiratory tract infection (19.4% and 17.5% of patients, respectively), headache (17.7% and 27.5%), diarrhea (14.5% and 7.5%), and abdominal pain (14.5% and 7.5%). See Table 1.
Complement 3 glomerulopathy.
The safety profile of Fabhalta is based on analysis of safety data in adult patients with native kidney C3G who received Fabhalta 200 mg twice daily (n = 38) or placebo (n = 36) in APPEAR-C3G for 26 weeks. The median duration of Fabhalta exposure was 6 months during the randomized period. The most commonly reported adverse drug reaction was upper respiratory tract infection (21%). See Table 2.
 Safety data in patients with recurrent C3G after kidney transplantation is limited. See Section 5.1, X2202 and roll-over extension study.
Safety data in patients with recurrent C3G after kidney transplantation is limited. See Section 5.1, X2202 and roll-over extension study.
Description of select adverse drug reactions. Infections.
In PNH clinical Phase 3 studies, 1 out of 102 PNH patients reported serious bacterial pneumonia while receiving treatment with Fabhalta. The patient, who was in APPOINT-PNH Phase 3 study, had been vaccinated against Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae type B and recovered following treatment with antibiotics while continuing treatment with Fabhalta.
In C3G phase 3 study (APPEAR-C3G), 1 patient had a bacteremia secondary to an encapsulated organism (S. pneumoniae) in the double-blind period prior a reported serious pneumococcal infection with pneumonia and sepsis while receiving treatment with Fabhalta in the open-label period. The patient had been vaccinated against Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae type B and recovered following treatment with antibiotics. Fabhalta treatment was interrupted and restarted after recovery.
Platelet count decreased in patients with PNH.
In patients with PNH, decreases in platelet counts were generally mild and transient. Some patients with concurrent anti-platelet antibodies or idiopathic bone marrow aplasia with pre-existing thrombocytopenia had further decreases to Grade 3 or 4 (based on CTCAE version 4.03).
Laboratory and vital signs.
Blood cholesterol and blood pressure increased in patients with PNH.
In APPLY-PNH and APPOINT-PNH, mean increases from baseline of approximately 37 mg/dL (0.952 mmol/L) and 17 mg/dL (0.433 mmol/L), respectively, were seen at month 6 for total cholesterol, and 32 mg/dL (0.830 mmol/L) and 18 mg/dL (0.467 mmol/L), respectively for LDL-cholesterol. The mean values remained within the normal ranges. Increases in blood pressure, particularly diastolic blood pressure (DBP), were also observed (mean increase 4.4 mmHg in APPLY-PNH and 3.4 mmHg in APPOINT-PNH at month 6). The mean DBP did not exceed 80 mmHg. Total cholesterol, LDL-C and DBP increases correlated with increases in haemoglobin (improvement in anaemia) in patients with PNH. The clinical relevance of such findings should be assessed based on individual patient characteristics and the patient should be managed accordingly.
In patients treated with iptacopan 200 mg twice a day in the C3G clinical study, no clinically relevant differences were observed in total cholesterol, LDL-cholesterol or blood pressure compared to placebo.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Limited data are available with regard to overdose in humans. During clinical studies, a few patients took up to 800 mg Fabhalta daily and this was well tolerated. In healthy volunteers, the highest dose was 1200 mg administered as a single dose and this was well tolerated.
General supportive measures and symptomatic treatment should be initiated in cases of suspected overdose.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
Pharmacotherapeutic group, ATC.
Pharmacotherapeutic group: complement inhibitors, ATC code: L04AJ08.
5.1 Pharmacodynamic Properties
Mechanism of action.
Iptacopan is a proximal complement inhibitor that targets Factor B (FB) to selectively inhibit the alternative pathway while leaving the direct signalling from the lectin and classical pathways intact. Inhibition of FB prevents the activity of alternative pathway related C3 convertase and the subsequent formation of C5 convertase.
In PNH, intravascular haemolysis (IVH) is mediated by the downstream membrane attack complex (MAC), while extravascular haemolysis (EVH) is facilitated by opsonisation with C3 fragments. Iptacopan acts proximally in the alternative pathway of the complement cascade to control both C3-mediated EVH and terminal complement-mediated IVH.
In C3G, overactivation of the alternative complement pathway leads to systemic C3 cleavage, resulting in C3 deposition and inflammation in the glomeruli, which are responsible for the pathogenesis of C3G and can lead to kidney damage and failure. By binding to Factor B, iptacopan selectively inhibits the effect of the alternative pathway.
Pharmacodynamics.
The onset of inhibition of the alternative complement pathway biomarkers, ex vivo alternative pathway assay and plasma Bb (fragment Bb of FB), was ≤ 2 hours after a single iptacopan dose in healthy volunteers.
In PNH patients receiving concomitant anti-C5 treatment and iptacopan 200 mg twice daily, the ex vivo alternative pathway assay and plasma Bb decreased from baseline by 54.1% and 56.1%, respectively, on the first observation on Day 8. In treatment-naïve PNH patients, these same biomarkers decreased from baseline by 78.4% and 58.9%, respectively, on the first observation after 4 weeks of treatment with iptacopan 200 mg twice daily.
In PNH patients on concomitant anti-C5 treatment and iptacopan 200 mg twice daily, the mean PNH red blood cells (RBC) clone size was 54.8% at baseline and increased to 89.2% after 13 weeks; the proportion of PNH Type II + III RBCs with C3 deposition was 12.4% at baseline and decreased to 0.2% after 13 weeks. In treatment-naïve PNH patients, the mean PNH RBC clone size was 49.1% at baseline and increased to 91.1% after 12 weeks; there were negligible PNH Type II + III RBCs with C3 deposition in this population due to the predominance of IVH.
Iptacopan reduces serum LDH levels. In PNH patients previously treated with eculizumab, all patients treated with iptacopan 200 mg twice daily achieved a reduction of LDH levels to < 1.5 times upper limit of normal (ULN) after 13 weeks and maintained the effect through the end of the study. In treatment-naïve PNH patients, iptacopan 200 mg twice daily reduced LDH by > 60% compared to baseline after 12 weeks and maintained the effect through the end of the study.
In C3G patients receiving Fabhalta 200 mg twice daily (APPEAR-C3G), the geometric mean serum C3 at baseline was 233 mg/L and increased to 798 mg/L at Day 14. Over this same period, the placebo group geometric mean serum C3 level decreased from 250 mg/L to 240 mg/L. At 6 months, the mean glomerular C3 deposition score (0-12) decreased by 0.8 (95% CI: -1.8, 0.3) from a baseline of 9.2 with Fabhalta and increased by 1.1 (95% CI: 0.1, 2.1) from a baseline of 9.6 with placebo. Also at 6 months, the geometric mean plasma soluble C5b-9 (also known as membrane attack complex (MAC)) and urine soluble C5b-9 decreased from baseline by 67% and 88%, respectively, compared to decreases of 3% and 36% in the placebo group, respectively.
Cardiac electrophysiology.
In a QTc clinical study in healthy volunteers, single supra-therapeutic iptacopan doses up to 1200 mg (which provided greater than 4-fold peak concentration of the MRHD) showed no effect on cardiac repolarisation or QT interval.
Clinical trials.
Paroxysmal nocturnal hemoglobinuria. The efficacy and safety of Fabhalta in adult patients with PNH were evaluated in two multi-centre, open-label, 24-week Phase 3 studies: an active comparator-controlled study (APPLY-PNH; NCT04558918) and a single arm study (APPOINT-PNH; NCT04820530).
APPLY-PNH: anti-C5 treatment experienced patients with PNH.
APPLY-PNH enrolled adult PNH patients with residual anaemia (haemoglobin < 10 g/dL) despite previous treatment with a stable regimen of anti-C5 treatment (either eculizumab or ravulizumab) for at least 6 months prior to randomization.
Ninety-seven patients were randomized in 8:5 ratio either to receive Fabhalta 200 mg orally twice daily (n=62) or to continue anti-C5 treatment (eculizumab n=23 or ravulizumab n=12) throughout the duration of the 24-week randomized controlled period (RCP). Randomization was stratified based on prior anti-C5 treatment and transfusion history within the last 6 months. Following completion of the 24-week RCP, all patients were eligible to enrol in a 24-week treatment extension period and receive Fabhalta monotherapy. Subsequently, patients were eligible to enter a separate long-term extension study.
Patients were required to be vaccinated against Neisseria meningitidis and recommended to be vaccinated against Streptococcus pneumoniae and Haemophilus influenzae type B. If the patient had not been previously vaccinated or if a booster was required, vaccination was administered at least 2 weeks prior to first dosing. If Fabhalta treatment was initiated earlier than 2 weeks after vaccination, antibacterial drug prophylaxis was administered.
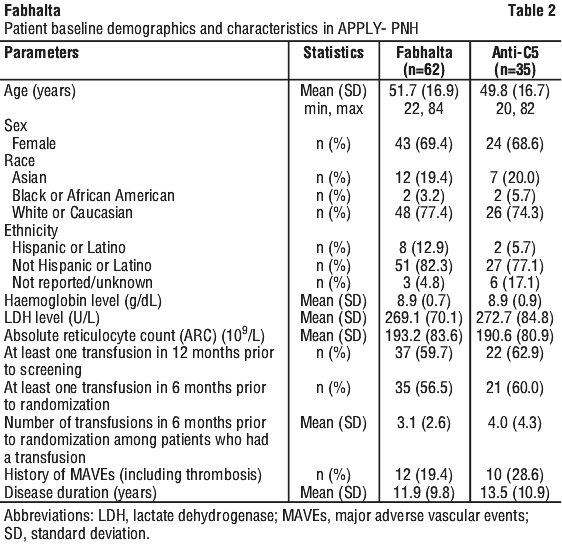
Demographics and baseline disease characteristics were generally well balanced between treatment groups (see Table 3). The mean time on prior anti-C5 treatment was 3.8 and 4.2 years for Fabhalta and anti-C5 groups, respectively. The baseline mean PNH RBC clone size (type II + III) was 64.6% for Fabhalta and 57.4% for the anti-C5 group. Mean baseline haemoglobin was 8.9 g/dL for both groups, with approximately 57% and 60% of patients requiring a transfusion in the 6 months prior to randomization, in the Fabhalta and anti-C5 groups, respectively. The mean baseline LDH level was 269.1 U/L for Fabhalta and 272.7 U/L for the anti-C5 group. There were 19.4% and 28.6% of patients with a history of MAVEs in the Fabhalta and anti-C5 groups, respectively.
During the RCP, one patient in the Fabhalta group discontinued treatment due to pregnancy; no patients in the anti-C5 group discontinued.
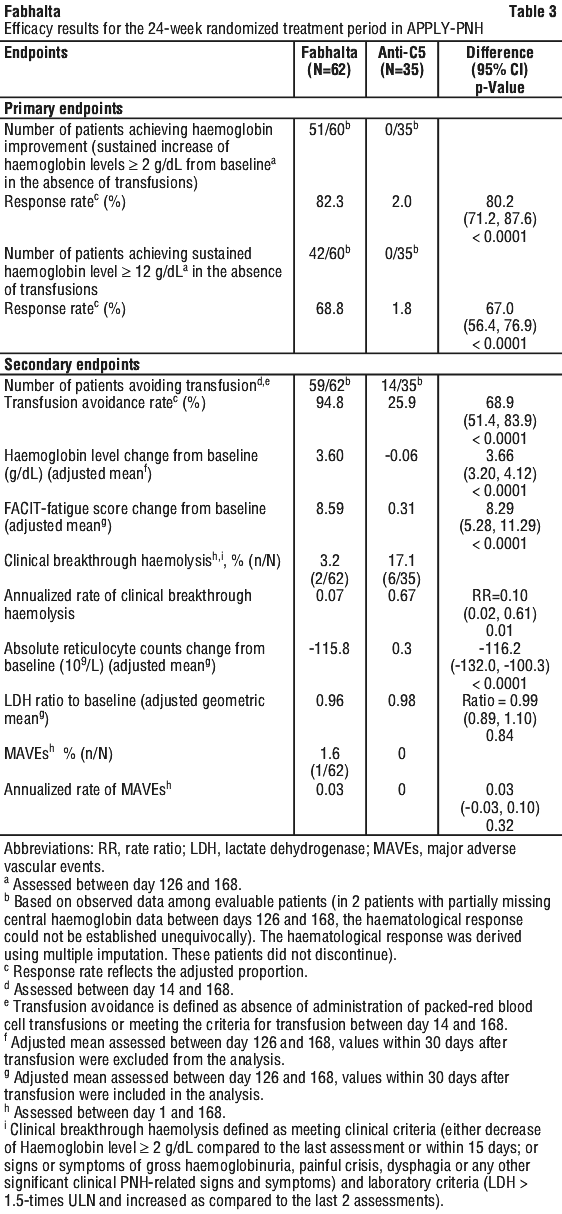 Efficacy was based on two primary endpoints to demonstrate superiority of Fabhalta to anti-C5 in achieving haematological response after 24 weeks of treatment, without a need for transfusion, by assessing the proportion of patients demonstrating: 1) sustained increase of ≥ 2 g/dL in haemoglobin levels from baseline (haemoglobin improvement) and/or 2) sustained haemoglobin levels ≥ 12 g/dL. Secondary endpoints included transfusion avoidance, change from baseline in haemoglobin levels, change from baseline in Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue scores, occurrence of clinical breakthrough haemolysis and change from baseline in absolute reticulocyte counts.
Efficacy was based on two primary endpoints to demonstrate superiority of Fabhalta to anti-C5 in achieving haematological response after 24 weeks of treatment, without a need for transfusion, by assessing the proportion of patients demonstrating: 1) sustained increase of ≥ 2 g/dL in haemoglobin levels from baseline (haemoglobin improvement) and/or 2) sustained haemoglobin levels ≥ 12 g/dL. Secondary endpoints included transfusion avoidance, change from baseline in haemoglobin levels, change from baseline in Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue scores, occurrence of clinical breakthrough haemolysis and change from baseline in absolute reticulocyte counts.
Fabhalta was superior to anti-C5 treatment, with a significant difference in response rate of 80.2% (82.3% vs 2%) for haemoglobin improvement (sustained increase of haemoglobin levels ≥ 2 g/dL from baseline) and 67% (68.8% vs 1.8%) for sustained haemoglobin level ≥ 12 g/dL without a need for RBC transfusion for both primary endpoints, after 24 weeks of treatment (p < 0.0001) (see Table 4).
Overall, more patients achieved haemoglobin improvement in the Fabhalta group (51/60) compared to the anti-C5 group (0/35), and sustained haemoglobin ≥ 12 g/dL (42/60 in the Fabhalta group compared to 0/35 in the anti-C5 group) without a need for RBC transfusion (see Table 4).
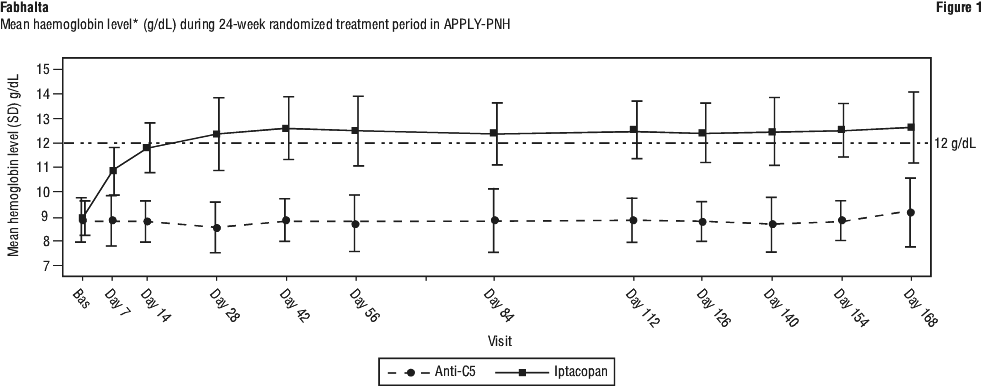
Fabhalta was also superior to anti-C5 treatment for transfusion avoidance rate with a treatment difference of 68.9% (94.8% vs 25.9% (p < 0.0001)) and change from baseline in haemoglobin level (treatment difference of +3.66 g/dL; p < 0.0001). The treatment effect of Fabhalta on haemoglobin was seen as early as day 7 and sustained during the study (see Figure 1).
Fabhalta was superior to anti-C5 treatment in improving fatigue as assessed by FACIT-Fatigue (treatment difference of +8.29 points; p < 0.001), and patients treated with Fabhalta experienced clinically meaningful improvements in patient reported fatigue from baseline (+8.59 points). Fabhalta was also superior to anti-C5 treatment in annualized rate of clinical breakthrough haemolysis (treatment difference of 90%; p=0.01) and reduction in absolute reticulocyte count from baseline (treatment difference of -116.2 x 109/L; p < 0.0001) consistent with the inhibition of EVH.
The LDH ratio to baseline was similar for both treatment groups, demonstrating that Fabhalta maintained control of IVH following discontinuation of anti-C5 treatment (see Table 4).

 The results for the primary endpoints were consistent across the predefined subgroups studied, including disease duration, age, sex, baseline haemoglobin, history of MAVEs, previous anti-C5 treatment (eculizumab or ravulizumab), the need for transfusion in the last 6 months, number of transfusions in the last 6 months (< 2 or ≥ 2), LDH level at baseline, and duration of previous anti-C5 treatment.
The results for the primary endpoints were consistent across the predefined subgroups studied, including disease duration, age, sex, baseline haemoglobin, history of MAVEs, previous anti-C5 treatment (eculizumab or ravulizumab), the need for transfusion in the last 6 months, number of transfusions in the last 6 months (< 2 or ≥ 2), LDH level at baseline, and duration of previous anti-C5 treatment.
APPOINT-PNH: complement inhibitor naïve study.
APPOINT-PNH was a single-arm study in 40 adult PNH patients (RBC clone size ≥ 10%) with haemoglobin < 10 g/dL and LDH > 1.5 ULN, who were not previously treated with a complement inhibitor. All 40 patients received Fabhalta 200 mg orally twice daily during the 24-week open-label core treatment period. Subsequently, patients were eligible to enrol in a 24-week treatment extension period and continue to receive Fabhalta, followed by a separate long-term extension study.
Patients were required to be vaccinated against Neisseria meningitidis and recommended to be vaccinated against Streptococcus pneumoniae and Haemophilus influenzae type B. If the patient had not been previously vaccinated or if a booster was required, vaccination was administered at least 2 weeks prior to or up to 2 weeks after the first dose. If Fabhalta treatment was initiated earlier than 2 weeks after vaccination, antibacterial drug prophylaxis treatment was administered.
Table 5 shows the patient baseline demographics and disease characteristics. No patients discontinued from the core treatment period of the study.
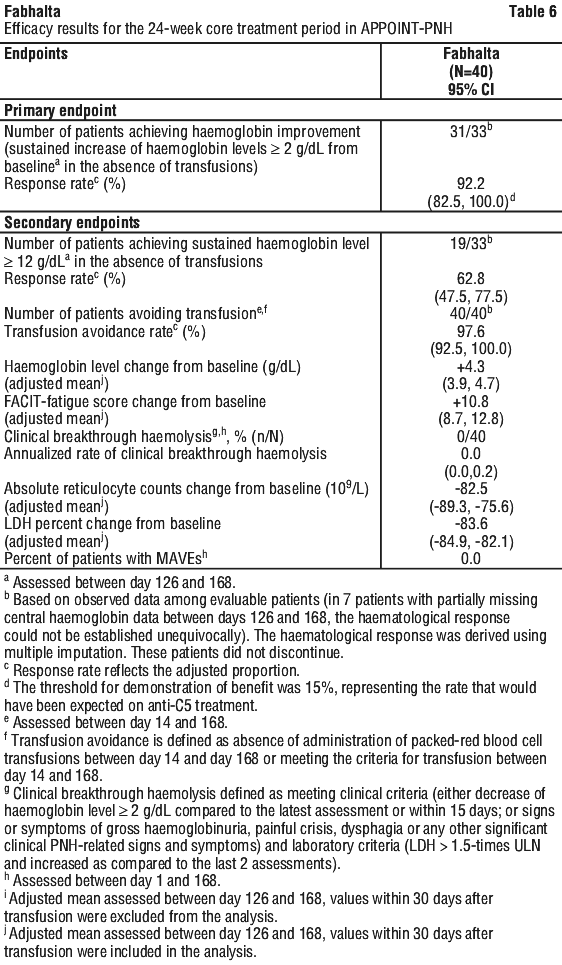
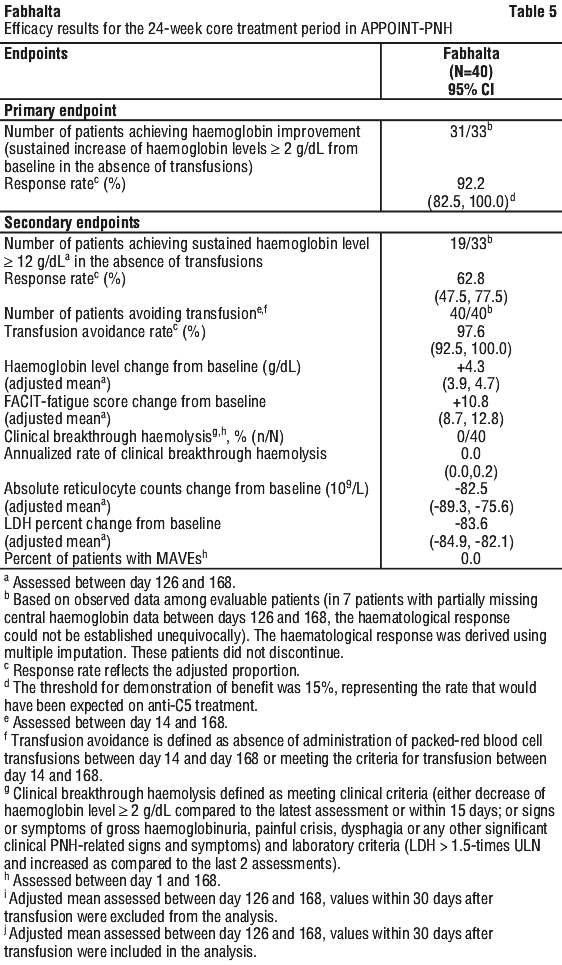 Efficacy was based on the primary endpoint assessing the effect of Fabhalta treatment on the proportion of patients achieving haemoglobin improvement (sustained increase of ≥ 2 g/dL in haemoglobin levels from baseline, without a need for RBC transfusion, after 24 weeks). Secondary endpoints included: sustained haemoglobin ≥ 12 g/dL (without a need for RBC transfusion) after 24 weeks, transfusion avoidance, change from baseline in haemoglobin levels, change from baseline in FACIT-Fatigue scores, occurrence of clinical breakthrough haemolysis and change from baseline in absolute reticulocyte counts.
Efficacy was based on the primary endpoint assessing the effect of Fabhalta treatment on the proportion of patients achieving haemoglobin improvement (sustained increase of ≥ 2 g/dL in haemoglobin levels from baseline, without a need for RBC transfusion, after 24 weeks). Secondary endpoints included: sustained haemoglobin ≥ 12 g/dL (without a need for RBC transfusion) after 24 weeks, transfusion avoidance, change from baseline in haemoglobin levels, change from baseline in FACIT-Fatigue scores, occurrence of clinical breakthrough haemolysis and change from baseline in absolute reticulocyte counts.
Fabhalta treatment resulted in a response rate of 92.2% (95% CI: 82.5, 100.0) for haemoglobin improvement, without a need for RBC transfusion, after 24 weeks. The response rate for patients achieving haemoglobin ≥ 12 g/dL, without a need for RBC transfusion was 62.8% (95% CI: 47.5, 77.5). Fabhalta treatment led to transfusion avoidance rate of 97.6% (95% CI: 92.5, 100.0). Patients treated with Fabhalta experienced clinically meaningful improvements in patient reported fatigue (FACIT-Fatigue score change from baseline +10.8; 95% CI: 8.7, 12.8). No patients experienced clinical breakthrough haemolysis or MAVEs. When compared to baseline, in patients treated with Fabhalta, haemoglobin levels increased by 4.3 g/dL (95% CI: 3.9, 4.7), absolute reticulocyte counts changed by -82.5 x 109/L (95% CI: -89.3, -75.6), and the LDH percent change was -83.6% (95% CI: -84.9, -82.1) after 24 weeks. The treatment effect of Fabhalta on LDH was seen as early as day 7 and reached < 1.5 ULN by day 14, which was sustained during the study. (See Table 6 and Figure 2).
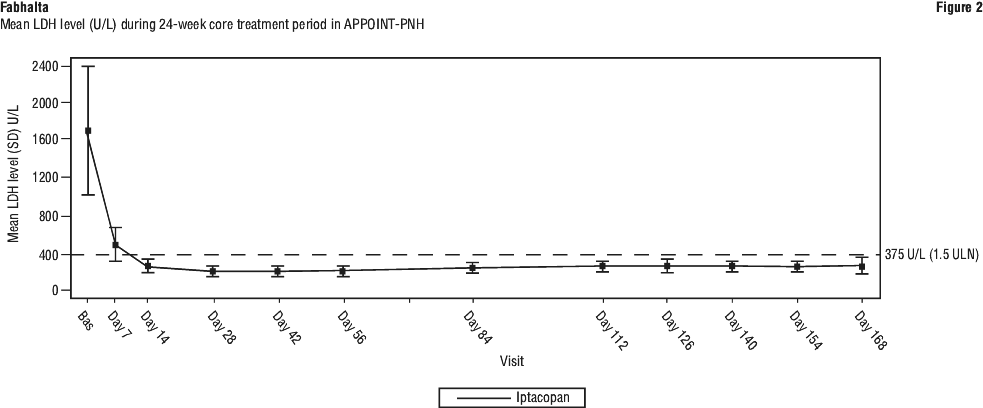 The results for the primary endpoint were consistent across the predefined subgroups examined, including disease duration, age, sex, baseline haemoglobin, history of MAVEs, need for transfusion in the last 6 months, and number of transfusions in the last 6 months (< 2 or ≥ 2).
The results for the primary endpoint were consistent across the predefined subgroups examined, including disease duration, age, sex, baseline haemoglobin, history of MAVEs, need for transfusion in the last 6 months, and number of transfusions in the last 6 months (< 2 or ≥ 2).
Complement 3 glomerulopathy. APPEAR-C3G: adult patients with native kidney C3G.
The efficacy and safety of Fabhalta in adult patients with C3G were evaluated in the multicentre, randomized, double-blind study (APPEAR-C3G). The study enrolled 74 adult patients with biopsy confirmed C3G. Patients who had a urine protein-to-creatinine ratio (UPCR) ≥ 1 g/g (or ≥ 0.113 g/mmol), an eGFR ≥ 30 mL/min/1.73 m2 and a reduced serum C3 were included. Patients with a transplanted kidney, rapidly progressive crescentic glomerulonephritis, kidney biopsy showing interstitial fibrosis/tubular atrophy of > 50% or monoclonal gammopathy of undetermined significance (MGUS) were excluded.
All patients were required to be on a maximally recommended or tolerated dose of an ACEI or ARB. Systemic corticosteroids up to 7.5 mg/day prednisone (or equivalent) and mycophenolate mofetil/sodium (MMF/MPS) of any dose were allowed. All of these therapies (i.e. ACEI/ARB, corticosteroids and MMF/MPS) were required to be at stable doses 90 days prior to randomization and throughout the study.
Patients were randomized (1:1) to receive either Fabhalta 200 mg orally twice daily (n=38) or placebo (n=36) for 6 months, followed by a 6-month open label treatment period in which all patients received Fabhalta 200 mg orally twice daily. Randomization was stratified according to whether or not patients were receiving concomitant immunosuppressive therapy (i.e. corticosteroid and/or MMF/MPS).
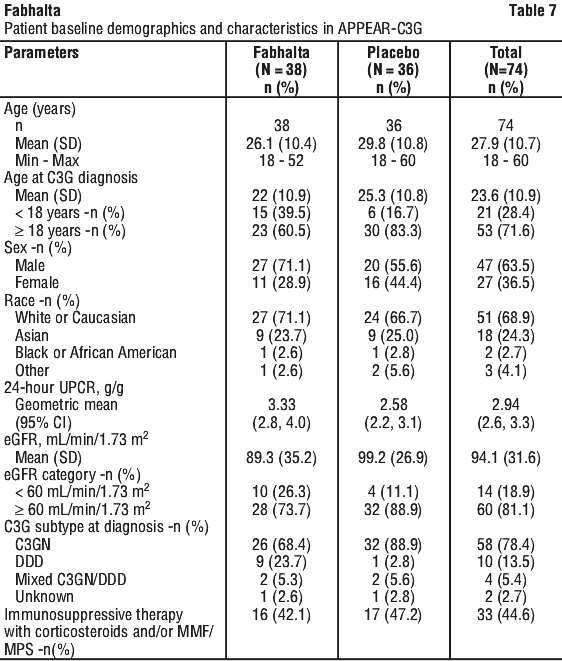
Baseline demographics and characteristics for the randomized patients are shown in Table 7. Overall, most (81.1%) patients had eGFR ≥ 60 mL/min/1.73 m2, and most had C3GN. The mean modelled historical eGFR slope prior to randomisation was -10.75 vs. -7.64 mL/min/1.73 m2 per year in iptacopan and placebo arms, respectively.
 The primary efficacy endpoint was percent change in 24-hour UPCR compared to baseline after 6 months of treatment.
The primary efficacy endpoint was percent change in 24-hour UPCR compared to baseline after 6 months of treatment.
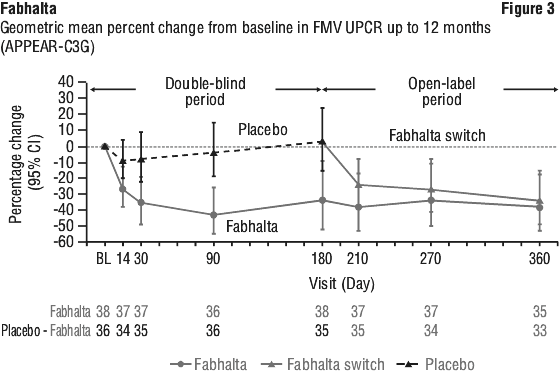
Fabhalta was superior to placebo. At month 6, there was a 30.2% reduction in proteinuria compared to baseline in the Fabhalta arm, compared to a 7.6% increase in the placebo arm (1-sided p=0.0014). See Table 8. 29.7% of patients in the Fabhalta arm achieved a ≥ 50% reduction in proteinuria by month 6, compared to 5.6% of the placebo arm.
Among patients initially randomised to Fabhalta, the reduction in 24-hour UPCR compared to baseline was 36.8% at month 12. Patients who switched from placebo to Fabhalta in the 6-month open label treatment period experienced a 31.0% reduction in 24-hour UPCR from Month 6 to Month 12. Descriptive results showing first morning void (FMV) UPCR trajectory is provided in Figure 3).

 Mean change in eGFR from baseline to month 6 was 1.3 and -0.9 mL/min/1.73 m2 for Fabhalta and placebo, respectively, resulting in a numerical difference of 2.2 mL/min/1.73 m2 (95% CI: -2.7, 7.1), which was not statistically significant. At 12 months, the mean change from baseline in eGFR was +0.44 mL/min/1.73 m2 (95% CI: -3.76, 4.64) in the iptacopan arm.
Mean change in eGFR from baseline to month 6 was 1.3 and -0.9 mL/min/1.73 m2 for Fabhalta and placebo, respectively, resulting in a numerical difference of 2.2 mL/min/1.73 m2 (95% CI: -2.7, 7.1), which was not statistically significant. At 12 months, the mean change from baseline in eGFR was +0.44 mL/min/1.73 m2 (95% CI: -3.76, 4.64) in the iptacopan arm.
X2202 and roll-over extension study.
X2202 was a single-arm, open-label Phase 2 study in adult patients with C3G in native kidney (n=16) and adult patients with recurrent C3G post-kidney transplantation (n=10). Patients continued on Fabhalta 200 mg twice daily in a roll-over extension study. Efficacy results are descriptive in nature.
C3G in native kidney.
In patients with native kidney (n=16), the mean baseline age was 26 years, mean eGFR was 70 mL/min/1.73 m2 and median 24-hour UPCR was 3.5 g/g. Mean UPCR and eGFR remained stable throughout the study. The mean change in eGFR from baseline was 6.5 and -11.1 mL/min/1.73 m2 on approximately Day 354 and 1164, respectively. Day 1164 data was missing in 3 patients who discontinued earlier.
Recurrent C3G post-kidney transplantation.
There is limited experience with the use of iptacopan in patients with recurrent C3G after transplantation.
Diagnosis of recurrent C3G required histological assessment of glomerular C3 staining intensity on a recent biopsy of the latest transplanted kidney. At baseline (n=10), the mean age was 36 years, mean eGFR was 53.9 mL/min/1.73 m2, median FMV UPCR was 0.08 g/g, and two patients had been transplanted following the loss of a first transplant. The mean time from last transplantation to first dose of iptacopan was 1.6 years. All patients were on MMF/MPS and/or corticosteroids in addition to calcineurin inhibitors.
In patients with recurrent C3G, iptacopan significantly reduced the histological C3 deposit score by 2.50 (p=0.0313) at 3 months. Effects on UPCR and eGFR were maintained for up to 39 months. Two patients discontinued after experiencing eGFR reductions of > 35 mL/min/1.73 m2 and had no data after discontinuation. In the remaining patients, the mean change in eGFR from baseline was -1.5 and -5.8 mL/min/1.73 m2 on approximately Day 354 (n=9) and 1164 (n=8), respectively.
5.2 Pharmacokinetic Properties
Absorption.
Following oral administration, iptacopan reached peak plasma concentrations approximately 2 hours post dose. At the recommended dosing regimen of 200 mg twice daily, steady-state is achieved in approximately 5 days with minor accumulation (1.4-fold). The Cmax and AUC data from a food-effect study involving administration of iptacopan to healthy volunteers under fasting conditions or with a high-fat meal indicated that exposure to iptacopan is not affected by food. Therefore, Fabhalta may be taken with or without food.
Distribution.
Iptacopan showed concentration-dependent plasma protein binding due to binding to the target FB in the systemic circulation. Iptacopan was 75% to 93% protein bound in vitro at relevant clinical plasma concentrations. After administration of iptacopan 200 mg twice daily, the apparent volume of distribution at steady state was approximately 288 L.
Metabolism.
Metabolism is a predominant elimination pathway for iptacopan with approximately 50% of the dose attributed to oxidative pathways. Metabolism of iptacopan includes N-dealkylation, O-deethylation, oxidation, and dehydrogenation, mostly driven by CYP2C8 (98%) with a small contribution from CYP2D6 (2%). Glucuronidation (UGT1A1, UGT1A3, UGT1A8) is a minor pathway. In plasma, iptacopan was the major component accounting for 83% of the AUC0-48hr. Two acyl glucuronides were the only metabolites detected in plasma and were minor, accounting for 8% and 5% of the AUC0-48hr. Iptacopan metabolites are not considered pharmacologically active.
Excretion.
In a human study, following a single 100 mg oral dose of [14C] iptacopan, mean total excretion of radioactivity (iptacopan and metabolites) was 71.5% in the faeces and 24.8% in the urine giving total mean excretion of > 96% of the dose. Specifically, 17.9% of the dose was excreted as parent iptacopan into the urine and 16.8% in faeces. The half-life (t1/2) of iptacopan at steady state is approximately 25 hours after administration of Fabhalta 200 mg twice daily.
Linearity/non-linearity.
At doses between of 25 mg and 200 mg twice daily, iptacopan was overall under dose proportional. However, oral doses of 100 mg and 200 mg were approximately dose proportional.
Special populations.
A population pharmacokinetic (PK) analysis was conducted on data from 234 patients. Age, body weight, eGFR, race and gender did not significantly influence iptacopan PK. Studies that included Asian subjects showed that the PK of iptacopan were similar to Caucasian (white) subjects.
Use in hepatic impairment.
Based on a study in subjects with mild (Child-Pugh A, n=8), moderate (Child-Pugh B, n=8) or severe (Child-Pugh C, n=6) hepatic impairment, a negligible effect on the exposure of iptacopan was observed compared to subjects with normal hepatic function. Unbound iptacopan Cmax increased 1.4-, 1.7- and 2.1-fold, and unbound iptacopan AUCinf increased by 1.5-, 1.6- and 3.7-fold in subjects with mild, moderate and severe hepatic impairment, respectively.
No dose adjustment is required for patients with mild or moderate hepatic impairment. The use of iptacopan is not recommended in patients with severe hepatic impairment (see Section 4.2 Dose and Method of Administration).
Use in renal impairment.
The effect of renal impairment on the clearance of iptacopan was assessed using a population pharmacokinetic analysis. There were no clinically relevant differences in the clearance of iptacopan between patients with normal renal function and patients with mild (eGFR 60 - < 90 mL/min/1.73 m2) or moderate (eGFR 30 - < 60 mL/min/1.73 m2) renal impairment, and no dose adjustment is required (see Section 4.2 Dose and Method of Administration). Patients with severe renal impairment or on dialysis have not been studied.
5.3 Preclinical Safety Data
Genotoxicity.
Iptacopan showed no genotoxicity in assays for bacterial reverse mutation (Ames test), chromosomal aberrations in vitro (in human lymphocytes) or in the in vivo rat peripheral blood micronucleus test.
Carcinogenicity.
The carcinogenic potential of iptacopan was investigated in a 6-month study in transgenic (Tg.rasH2) mice and in a 2-year study in rats, both conducted by the oral route. Iptacopan was not carcinogenic in either species up to the highest doses tested (1000 mg/kg/day in mice and 750 mg/kg/day in rats). These doses yield exposure to iptacopan 4.4-times higher in mice (based on plasma AUC for total drug) and 44-times higher in rats (based on unbound AUC) than in patients at the MRHD.6 Pharmaceutical Particulars
6.1 List of Excipients
Capsule fill: none.
Capsule shell: hard gelatin, red iron oxide (E 172), titanium dioxide (E 171), and yellow iron oxide (E 172).
Printing ink: black iron oxide (E 172), concentrated ammonia solution (E 527), propylene glycol (E 1520), potassium hydroxide (E 525), and shellac (E 904).
6.2 Incompatibilities
Fabhalta is not anticipated to have clinically relevant interactions with other drug products (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
Fabhalta must be kept out of the reach and sight of children.
6.5 Nature and Contents of Container
Pack of 56 hard capsules in PVC/PE/PVdC (triplex) blister packs backed with aluminium foil.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
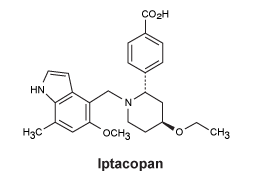
Iptacopan hydrochloride monohydrate has a molecular formula C25H30N2O4.HCl.H2O and a molecular mass of 477.00. It is a powder with pKa values of 8.9 and 3.7 and pH-dependant solubility.
Chemical structure.
CAS number.
1644670-37-0.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription medicine.
Summary Table of Changes