1 Name of Medicine
Benralizumab.
2 Qualitative and Quantitative Composition
Each Fasenra prefilled syringe contains 30 mg benralizumab in 1 mL (30 mg/mL).
Each Fasenra Pen prefilled pen contains 30 mg benralizumab in 1 mL (30 mg/mL).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection.
Clear to opalescent, colourless to yellow solution for injection in a prefilled syringe (Fasenra) or prefilled pen (Fasenra Pen).
4.1 Therapeutic Indications
Asthma.
Fasenra is indicated as add-on therapy in patients aged 12 years and over with severe eosinophilic asthma (blood eosinophil count ≥ 300 cells/microL or ≥ 150 cells/microL if on oral corticosteroid treatment) (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Relapsed or refractory eosinophilic granulomatosis with polyangiitis (EGPA).
Fasenra is indicated as an add-on treatment for relapsing or refractory eosinophilic granulomatosis with polyangiitis (EGPA) in adult patients aged 18 years and over (see Section 5.1 Pharmacodynamic Properties, Clinical trials).4.2 Dose and Method of Administration
Fasenra should be prescribed by a health care professional in consultation with a specialist physician experienced in the diagnosis and treatment of conditions for which Fasenra is indicated (see Section 4.1 Therapeutic Indications).
Fasenra is intended for long-term treatment. A decision to continue therapy should be made at least annually based on disease severity and level of disease control.
Asthma.
Treatment with high-dose inhaled corticosteroids (ICS) and long-acting β-agonists (LABA) should be optimised prior to commencement of treatment with Fasenra.
Adults and adolescents (12 years and over).
The recommended dose is 30 mg of Fasenra by subcutaneous injection every 4 weeks for the first 3 doses, and then every 8 weeks thereafter.
EGPA.
The recommended dosage is 30 mg of Fasenra administered by subcutaneous injection every 4 weeks.
Missed dose.
If an injection is missed on a planned date, dosing should resume as soon as possible on the indicated regimen; a double dose must not be administered.
Special patient populations.
Renal impairment.
No dose adjustment is required for patients with renal impairment (see Section 5.2 Pharmacokinetic Properties, Special patient populations, Renal impairment).
Hepatic impairment.
No dose adjustment is required for patients with hepatic impairment (see Section 5.2 Pharmacokinetic Properties, Special patient populations, Hepatic impairment).
Use in the elderly.
No dose adjustment is required for elderly patients (see Section 5.2 Pharmacokinetic Properties, Special patient populations, Elderly (≥ 65 years old)).
Paediatric use.
The safety and efficacy of Fasenra in children with asthma below 12 years of age have not been established.
The safety and efficacy of Fasenra in children and adolescents (below 18 years of age) with EGPA have not been established.
Method of administration.
Fasenra is for single use in one patient only. Discard any residue.
Fasenra is administered as a subcutaneous injection.
Administer Fasenra into the thigh or abdomen. If somebody else administers the injection, the upper arm can also be used. Do not administer into areas where the skin is tender, bruised, erythematous, or hardened.
Instructions for administration.
Do not shake. Do not use if frozen.
Prior to administration, if the Fasenra prefilled syringe/Fasenra Pen has been in the refrigerator allow Fasenra to reach room temperature (approximately 30 minutes).
Visually inspect Fasenra for particulate matter and discolouration prior to administration. Fasenra is clear, colourless to yellow, and may contain translucent or white to off white particles. Do not use Fasenra if liquid is cloudy, discoloured, or if it contains large particles or foreign particulate matter.
Additional information and instructions for the preparation and administration of Fasenra using the prefilled syringe or prefilled pen (Fasenra Pen) are given in the Instructions for Use booklet provided in each pack.4.3 Contraindications
Fasenra is contraindicated in patients who have known hypersensitivity to benralizumab or any of its excipients (see Section 4.4 Special Warnings and Precautions for Use, Hypersensitivity reactions).
4.4 Special Warnings and Precautions for Use
Fasenra should not be used to treat acute asthma exacerbations.
Patients should be instructed to seek medical advice if their asthma remains uncontrolled or worsens after initiation of treatment.
Reduction in oral corticosteroids (OCS) dose, if appropriate, should be gradual and performed under the supervision of a physician. Abrupt discontinuation of OCS after initiation of Fasenra therapy is not recommended.
Hypersensitivity reactions.
Hypersensitivity reactions (e.g. anaphylaxis, angioedema, urticaria, urticaria papular, rash) have occurred following administration of Fasenra (see Section 4.8 Adverse Effects (Undesirable Effects)). These reactions may occur within hours of administration, but in some instances have a delayed onset (i.e. days). A history of serious hypersensitivity/anaphylaxis unrelated to benralizumab may be a risk factor for anaphylaxis following Fasenra administration.
In the event of a hypersensitivity reaction, Fasenra should be discontinued and appropriate therapy initiated.
Parasitic (helminth) infection.
Eosinophils may be involved in the immunological response to some helminth infections. Patients with known helminth infections were excluded from participation in clinical trials. It is unknown if Fasenra may influence a patient's response against helminth infections.
Treat patients with pre-existing helminth infections before initiating therapy with Fasenra. If patients become infected while receiving treatment with Fasenra and do not respond to anti-helminth treatment, discontinue treatment with Fasenra until infection resolves.
Use in the elderly.
No dose adjustment is required for elderly patients (see Section 4.2 Dose and Method of Administration, Special patient populations, Use in the elderly; Section 5.2 Pharmacokinetic Properties, Special patient populations, Elderly (≥ 65 years old)).
Paediatric use.
The safety and efficacy of Fasenra in children below 12 years of age have not been established.
In Phase 3 studies SIROCCO/CALIMA, the treatment responses in adolescent patients (12 to 17 years of age) were less than that observed in adults, however they were not powered to detect a response in this sub-group. The adverse event profile in adolescents was generally similar to the overall population in these studies of up to 56 weeks duration as well as in a 108-week extension study (BORA).
Effects on laboratory tests.
As expected based on the mechanism of action of benralizumab, in the pivotal Phase 3 trials, following subcutaneous administration of benralizumab at the recommended dose blood eosinophils were reduced to a median absolute blood eosinophil count of 0 cells/microL. This magnitude of reduction was seen at the first observed time point, 4 weeks of treatment, and was maintained throughout the treatment period. Basophils also express IL-5Rα, and were also reduced.4.5 Interactions with Other Medicines and Other Forms of Interactions
In a randomized, double-blind parallel-group study of 103 patients aged between 12 and 21 years with severe asthma, the humoral antibody responses induced by seasonal influenza virus vaccination were similar between benralizumab 30 mg and placebo. Vaccine efficacy was not directly assessed in this study.
Cytochrome P450 enzymes, efflux pumps and protein-binding mechanisms are not involved in the clearance of benralizumab. There is no evidence of IL-5Rα expression on hepatocytes. Eosinophil depletion does not produce chronic systemic alterations of proinflammatory cytokines.
An effect of benralizumab on the pharmacokinetics of co-administered medications is not expected. Based on population pharmacokinetic analysis, commonly co-administered medications had no effect on the benralizumab clearance in patients with asthma.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No fertility studies have been conducted with benralizumab in humans or animals.
Examination of surrogate fertility parameters (including organ weights and histopathology of reproductive tissues) in male and female cynomolgus monkeys treated with benralizumab at intravenous doses up to 25 mg/kg or at subcutaneous doses of up to 30 mg/kg once every 2 weeks for 9 months suggested no impairment of fertility. Systemic exposure (serum AUC) at the no adverse effect dose in monkeys was more than 400 times that in patients at the maximum recommended human dose.
(Category B1)
It is preferable to avoid the use of Fasenra during pregnancy, especially during the third trimester due to the potential for eosinophil depletion in the newborn. Administration of Fasenra to pregnant women should only be considered if the expected benefit to the mother is greater than any possible risk to the fetus.
IgG antibodies such as benralizumab are increasingly transported across the placenta as pregnancy progresses; therefore, greater fetal exposure occurs in the third trimester of pregnancy.
The effect of Fasenra on human pregnancy is unknown.
No adverse effects on pre- and post-natal survival, growth or development were observed in cynomolgus monkeys with maternal administration of intravenous doses of benralizumab (10 or 30 mg/kg) once every 2 weeks from early pregnancy (gestation day 20-22) to 1-month post-partum. Serum benralizumab levels in infants were 66% of maternal levels 7 days post-partum and declined over time (e.g. 10% of maternal levels 3 months post-partum). While there was no effect observed on the primary and secondary humoral immune responses to immunisation or levels of serum IgM, IgG and IgA in the offspring of the treated monkeys, there was a marked eosinophil depletion, consistent with significant placental transfer of benralizumab that resulted in pharmacologically active drug levels in infants. The immunological response to parasitic infection was not examined in the offspring.
It is unknown whether benralizumab is excreted in human milk. Since antibodies can be secreted in human milk a risk to the breastfed child cannot be excluded.
A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from benralizumab therapy taking into account the benefit of breast-feeding for the child and the benefit of therapy for the mother.4.7 Effects on Ability to Drive and Use Machines
Fasenra has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Clinical trials experience - asthma.
A total of 895 patients received the recommended dose of Fasenra (30 mg every 4 weeks for the first 3 doses, and then every 8 weeks thereafter) within the 3 placebo-controlled Phase 3 clinical trials - 822 patients (including 38 adolescents) in SIROCCO and CALIMA (48 and 56-week duration respectively) and 73 patients (adults only) in a 28-week OCS sparing trial (ZONDA). Note - all 3 trials assessed 2 dosing regimens of Fasenra compared to placebo, however only the safety data for the recommended dosing regimen (Q8w; see Section 4.2 Dose and Method of Administration) has been presented below.
In clinical trials in patients with severe asthma with eosinophilic phenotype the most commonly reported adverse drug reactions during treatment were headache and pharyngitis.
Phase 3 exacerbation trials (SIROCCO and CALIMA). Table 1 presents the most common (≥ 3% frequency) adverse events regardless of causality from the two placebo-controlled trials (SIROCCO/CALIMA) in patients receiving the recommended dose (Q8w) of benralizumab. Table 2 presents the SIROCCO/CALIMA adverse drug reactions reported at a frequency less than 3%. All patients (including placebo) were on a background of high-dose ICS/LABA.


Adolescents (12 to 17 years of age).
The frequency, type and severity of adverse drug reactions in the adolescent population were observed to be similar to those seen in adults.
Phase 3 OCS sparing trial (ZONDA). In ZONDA all patients were taking daily OCS (7.5 to 40 mg per day) in addition to regular use of high-dose ICS/LABA. In general, the safety results in ZONDA were similar to those observed in SIROCCO and CALIMA.
Long-term safety. In a 56-week double blind, randomized, parallel-group extension trial (BORA) in patients with asthma from SIROCCO, CALIMA and ZONDA trials, 842 patients were treated with Fasenra at the recommended dose and remained in the trial. The overall adverse event profile was similar to the asthma trials described above. Additionally, in an open-label safety extension trial (MELTEMI) in patients with asthma from previous trials, 226 adult patients were treated with Fasenra at the recommended dose for up to 42.3 months. Combined with the treatment period in previous studies, this corresponds to median follow-up of 3.4 years (range 8.5 months - 5.3 years). The safety profile during this follow-up period was consistent with the known safety profile of Fasenra.
Adolescent patients aged 12 to 17 (n=61) from SIROCCO and CALIMA continued treatment with Fasenra (at the recommended dose) in BORA for up to 108 weeks. Safety was consistent with the predecessor trials.
Clinical trials experience - EGPA.
The safety profile of a total of 70 patients with EGPA, who received Fasenra 30 mg every 4 weeks during an active-controlled Phase 3 clinical study (MANDARA) of 52 weeks duration, was similar to the established safety profile of Fasenra. This incidence of adverse reactions were similar to those reported in asthma, with the exception of headache, which occurred in 17% of Fasenra treated patients compared to 8% in asthma. No additional adverse reactions were identified.
Immunogenicity. Treatment with benralizumab, like other monoclonal antibodies, may result in an anti-drug antibody (ADA) response (see Section 5.1 Pharmacodynamic Properties, Immunogenicity). However, there is no apparent correlation of ADA development to efficacy or adverse events.
Post marketing experience.
In addition to the above, Table 3 describes adverse reactions that have been identified during post-approval use of Fasenra.

Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Doses of up to 200 mg were administered subcutaneously in clinical trials to patients with eosinophilic disease without evidence of dose-related toxicities.
There is no specific treatment for an overdose with benralizumab. If overdose occurs, the patient should be treated supportively with appropriate monitoring as necessary.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Benralizumab is an antibody that binds to the alpha subunit of the human interleukin-5 receptor (IL-5Rα) with high affinity (16 picoM) and specificity. The IL-5 receptor is specifically expressed on the surface of eosinophils and basophils. The absence of fucose sugar units in the Fc domain of benralizumab results in high affinity (45.5 nanoM) for FcγRIII receptors on immune effectors cells such as natural killer (NK) cells leading to apoptosis of eosinophils and basophils through enhanced antibody-dependent cell-mediated cytotoxicity (ADCC).
Eosinophilic inflammation is an important component in the pathogenesis of asthma. Eosinophils are a rich source of proinflammatory mediators (e.g. eicosanoids, leukotrienes, cytokines) and granule proteins (e.g. eosinophil cationic protein, eosinophil peroxidase, eosinophil neurotoxin and major basic protein). Benralizumab, by enhanced ADCC, reduces eosinophilic inflammation.
Pharmacodynamic effects.
The pharmacodynamic response (blood eosinophil depletion) following repeat subcutaneous dosing was evaluated in asthma patients in a 12-week Phase 2 trial. Patients with mild-moderate asthma received 1 of 3 doses of benralizumab [25 mg (n=7), 100 mg (n=6) or 200 mg (n=6) subcutaneous] or placebo (n=6) every 4 weeks for a total of 3 doses. Median blood eosinophil levels at baseline were 400, 200, 120 and 200 cells/microL in the 25, 100, and 200 mg benralizumab and placebo groups, respectively. Blood eosinophil depletion was observed following subcutaneous administration of benralizumab at all dose levels and no depletion was observed in the placebo group. Twenty-four hours post-dosing, all benralizumab dosage groups demonstrated complete or near complete depletion of median blood eosinophil levels (0, 0 and 5 cells/microL respectively). There were no changes in median blood eosinophils in the placebo group. The effect on blood eosinophil depletion was maintained throughout the dosing period.
In a Phase 1 trial, the effect of benralizumab on eosinophils in airway mucosa was evaluated in asthmatic patients with 2.5% or more eosinophils in sputum. Patients received 100 or 200 mg subcutaneous benralizumab once every 4 weeks for 8 weeks (total benralizumab subcutaneous group n=9) or matching placebo (n=5). At the end of the 12-week treatment period, there was a median reduction from baseline in eosinophils in the airway mucosa of 96% in the total benralizumab subcutaneous group compared to a 47% reduction from baseline in the placebo group which was statistically significant (p=0.039).
In the Phase 1 trial, treatment with benralizumab was also associated with reductions in blood basophils, and in both Phase 1 and 2 trials eosinophil granule products such as serum eosinophil derived neurotoxin (EDN) and eosinophil cationic protein (ECP).
In the two pivotal Phase 3 asthma trials (SIROCCO and CALIMA), following subcutaneous administration of benralizumab at the recommended dose blood eosinophils were reduced to a median absolute blood eosinophil count of 0 cells/microL, which corresponds to a median reduction of 100% (see Section 5.1 Pharmacodynamic Properties, Clinical trials). This magnitude of reduction was seen at the first observed time point, 4 weeks of treatment, and was maintained throughout the treatment period. Maintenance of eosinophil depletion was observed throughout the 56-week extension phase (BORA extension trial) consistent with previous trials.
In patients with EGPA, depletion of blood eosinophils was consistent with the effect observed in the asthma trials. Blood eosinophil depletion was seen at the first observed time point, 1 week of treatment, and was maintained throughout the 52-week treatment period.
Immunogenicity.
Overall, treatment-emergent anti-drug antibody (ADA) response developed in 107 out of 809 (13%) of asthma patients treated with Fasenra at the recommended dosing regimen during the 48 to 56-week treatment period. In a majority of the ADA positive patients, in vitro neutralizing antibodies were detected. Anti-benralizumab antibodies were associated with increased clearance of benralizumab and increased blood eosinophil levels in patients with high ADA titers compared to antibody negative patients. No evidence of an association of ADA with efficacy or safety was observed.
Following a second year of treatment of these asthma patients from the Phase 3 placebo-controlled trials, an additional 18 out of 510 (4%) had newly developed treatment-emergent antibodies. Overall, in patients who were ADA positive in the predecessor trials, titers remained stable or declined in the second year of treatment. Consistent with predecessor trials, no evidence of an association of ADA with efficacy or safety was observed.
In patients with EGPA, treatment-emergent ADA response developed in 6 out of 67 (9%) patients treated with Fasenra during the Phase 3 active-controlled 52-weeks treatment period. Neutralising antibody activity was detected in one of the ADA positive patients.
The data reflect the percentage of patients whose test results were positive for antibodies to benralizumab in specific assays. The observed treatment-emergent ADA response is highly dependent on several factors, including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medication, and underlying disease. For these reasons, comparison of the treatment-emergent ADA response to benralizumab with the treatment-emergent ADA response to other products may be misleading.
Clinical trials.
Severe asthma.
The safety and efficacy of Fasenra as an add-on therapy in patients with severe asthma were evaluated in 3 randomised, double-blind, parallel-group, placebo-controlled Phase 3 clinical trials:
two replicate long-term exacerbation trials in adults and adolescents (12 years and older) with 48 and 56 weeks duration (SIROCCO and CALIMA respectively); and
one 28-week OCS reduction trial in adults (18 years and over (ZONDA)).
While all 3 trials assessed 2 dosing regimens compared to placebo, the recommended dosing regimen is Fasenra administered every 4 weeks for the first 3 doses, then every 8 weeks thereafter (referred herein as Q8w; see Section 4.2 Dose and Method of Administration) as no additional benefit was observed with the more frequent dosing regimen (administered every 4 weeks). Only the results for the recommended Q8w dosing regimen have been presented below.
A total of 805, 881 and 148 patients were randomised to treatment with Fasenra Q8w and placebo in SIROCCO, CALIMA and ZONDA respectively. This included 84 adolescents (SIROCCO/CALIMA combined; 38 within the Fasenra Q8w arms and 4 within the placebo arms). See Table 4 for further details.

 SIROCCO and CALIMA. Patients enrolled into SIROCCO and CALIMA were required to have a history of ≥ 2 asthma exacerbations requiring oral or systemic corticosteroid treatment in the past 12 months, an Asthma Control Questionnaire-6 (ACQ-6) score of ≥ 1.5 at screening and reduced lung function at baseline [pre-bronchodilator forced expiratory volume in 1 second (FEV1) < 80% in adults and < 90% in adolescents] despite treatment with high dose ICS (SIROCCO) or with medium- or high-dose ICS (CALIMA) and their current standard of care. Medium- and high-dose ICS were defined as ≥ 250 microgram and ≥ 500 microgram/day fluticasone propionate dry powder formulation or equivalent respectively. The medium-dose ICS arm in CALIMA was assessed as a descriptive analysis only and has not been discussed further.
SIROCCO and CALIMA. Patients enrolled into SIROCCO and CALIMA were required to have a history of ≥ 2 asthma exacerbations requiring oral or systemic corticosteroid treatment in the past 12 months, an Asthma Control Questionnaire-6 (ACQ-6) score of ≥ 1.5 at screening and reduced lung function at baseline [pre-bronchodilator forced expiratory volume in 1 second (FEV1) < 80% in adults and < 90% in adolescents] despite treatment with high dose ICS (SIROCCO) or with medium- or high-dose ICS (CALIMA) and their current standard of care. Medium- and high-dose ICS were defined as ≥ 250 microgram and ≥ 500 microgram/day fluticasone propionate dry powder formulation or equivalent respectively. The medium-dose ICS arm in CALIMA was assessed as a descriptive analysis only and has not been discussed further.
Patients were stratified 2:1 according to baseline blood eosinophils count (≥ 300 or < 300 cells/microL). The primary efficacy population (intent-to-treat (ITT)) in both studies was patients with a high-dose ICS/LABA and a baseline blood eosinophil count ≥ 300 cells/microL. Prespecified analyses were also conducted on the high-dose ICS/LABA populations with a baseline blood eosinophil count < 300 cells/microL (patients not included within the ITT population) and the full analysis set (FAS) for predefined eosinophil ranges. Table 5 provides a summary of the randomised patient numbers for the different analysis groups.
ITT population (high-dose ICS/LABA and baseline blood eosinophil count ≥ 300 cells/microL).
The primary endpoint for both trials was the annual asthma exacerbation rate ratio versus placebo within the primary efficacy population (ITT). Key secondary endpoints were FEV1 and total asthma symptom score. Other lung function, symptom control and quality of life measures were also assessed.
Demographic, key respiratory and other baseline disease characteristics were balanced across the ITT treatment groups (Fasenra Q8w and placebo) in both trials. The demographic and patient characteristics in the ITT population were generally similar to the overall population for the two treatment arms, as well as the < 300 cells/microL subgroup.
Treatment with Fasenra Q8w significantly reduced the annual rate of exacerbations (primary endpoint) by 51% (SIROCCO) and 28% (CALIMA) compared to placebo (see Table 6). Similar reductions were observed compared to placebo for exacerbations requiring hospitalisations only (52%) and emergency room visits only (77%) in SIROCCO. In CALIMA, there were too few events in the placebo treatment arm to draw conclusions for exacerbations requiring hospitalisation or emergency room visits.
Clinically and statistically significant improvements in lung function were also observed with a 0.159 L (SIROCCO) and 0.116 L (CALIMA) increase in pre-bronchodilator FEV1 relative to placebo (see Table 6). Compared with placebo, Fasenra Q8w provided consistent improvements over time in the mean change from baseline in FEV1. Benefits of Fasenra Q8w on lung function were further supported by improvements in the mean change for morning and evening peak expiratory flow (PEF) from baseline compared to placebo.
Significant improvements were also observed for total asthma symptoms score and the Asthma Control Questionnaire (ACQ-6) after treatment with Fasenra Q8w relative to placebo (see Table 6). Significant improvements in quality of life, as measured by the Standardised Asthma Quality of Life Questionnaire for 12 years and older (AQLQ(S)+12), were also demonstrated for Fasenra Q8w compared to placebo (see Table 6).

Baseline blood eosinophil subgroup analyses.
While efficacy benefits were observed with Fasenra compared to placebo irrespective of baseline eosinophil count, increasing baseline eosinophil counts were identified as a potential predictor of improved treatment response (see Figure 1).

Prior exacerbation history.
A subgroup analysis on prior exacerbation history within the ITT population indicated that a higher prior exacerbation history may also be a potential predictor of an improved treatment response. When considered alone, or in combination with a higher baseline eosinophil count, these factors may further identify patients who may achieve a greater response from treatment with Fasenra.
OCS dose reduction trials (ZONDA and PONENTE). ZONDA, a placebo-controlled study, and PONENTE, an open-label study, evaluated the effect of Fasenra on reducing the use of maintenance OCS.
ZONDA.
ZONDA included patients who were treated with daily OCS (7.5 to 40 mg/day) in addition to regular use of high-dose ICS/LABA with or without additional controller(s) to maintain asthma control. The definition of high-dose ICS was as per SIROCCO/CALIMA. There was an 8-week run-in period during which a patient's OCS dose was titrated to the minimum effective dose while maintaining asthma control. The baseline median OCS dose was 10 mg (range: 8-40 mg) for both treatment groups. Patients were also required to have blood eosinophil counts ≥ 150 cells/microL and a history of at least one exacerbation in the past 12 months. All patients were included in the analysis, including 20 patients with a baseline blood eosinophil count of ≥ 150-299 cells/microL (12 in Fasenra Q8w arm and 11 in the placebo arm).
Demographic, key respiratory and other baseline disease characteristics were balanced across the treatment groups (Fasenra Q8w and placebo).
The primary endpoint was percent (%) reduction from baseline of the final OCS dose during Weeks 24 to 28, while maintaining asthma control. Compared to placebo, patients receiving Fasenra Q8w achieved greater reductions in daily maintenance OCS dose while still maintaining asthma control. Reductions of ≥ 50% in the OCS dose were observed in 66% of patients receiving Fasenra Q8w compared to 37% for the placebo arm (see Table 7). The proportion of patients with a mean final OCS dose ≤ 5 mg at Weeks 24 to 28 were 59% for Fasenra Q8w and 33% for placebo (odds ratio 2.74 (95% CI: 1.41, 5.31), p=0.002). Patients with an optimized baseline OCS dose (12.5 mg or less) were eligible to achieve a 100% reduction in OCS dose during the study. A significant difference was observed in the percentage of eligible patients who achieved 100% reduction with Fasenra Q8w compared to placebo (52.4% vs 19.0%; Odds ratio 4.19 (95% CI: 1.58, 11.12), p=0.002).
 Fasenra Q8w demonstrated a 70% reduction in the annual asthma exacerbation rate over 28 weeks compared with placebo (Rate ratio: 0.30 (95% CI: 0.17, 0.53), p < 0.001), and a 93% reduction in the annual asthma exacerbation rate associated with an emergency room visit or hospitalisations (Rate ratio: 0.07 (95% CI: 0.01, 0.63), p = 0.018) over 28 weeks compared with placebo.
Fasenra Q8w demonstrated a 70% reduction in the annual asthma exacerbation rate over 28 weeks compared with placebo (Rate ratio: 0.30 (95% CI: 0.17, 0.53), p < 0.001), and a 93% reduction in the annual asthma exacerbation rate associated with an emergency room visit or hospitalisations (Rate ratio: 0.07 (95% CI: 0.01, 0.63), p = 0.018) over 28 weeks compared with placebo.
PONENTE.
PONENTE enrolled 598 adult patients with severe asthma (blood eosinophil count ≥ 150 cells/microL at entry or ≥ 300 cells/microL in the past 12 months if study entry count was < 150 cells/microL) who were OCS-dependent. The primary endpoints included the proportion of patients who achieved a final OCS dose less than or equal to 5 mg while maintaining asthma control and taking into account adrenal function. The proportion of patients who achieved an OCS dose less than or equal to 5 mg (while maintaining asthma control and not limited by adrenal function) was 81.9%. Effects on OCS reduction were similar irrespective of blood eosinophil count at study entry (including patients with blood eosinophils < 150 cells/microL) and maintained over an additional period of 24 to 32 weeks.
Long term data. The long-term safety and efficacy of Fasenra was evaluated in a double-blind, randomised, parallel group, Phase 3, 56-week extension trial (BORA) in adult and adolescent (aged 12 years and older) patients from the predecessor studies - SIROCCO, CALIMA and ZONDA. The long-term safety of Fasenra was evaluated in an open-label safety extension trial MELTEMI (see Section 4.8 Adverse Effects (Undesirable Effects)).
BORA assessed the long-term effect of Fasenra on annual asthma exacerbation rate, lung function and ACQ-6, AQLQ(S)+12 at the 2 dosing regimens studied in the predecessor studies.
A total of 543 adult and adolescent patients were randomised to receive Fasenra Q8w in the predecessor trials and were treated and remained in the BORA extension trial - 512 from SIROCCO/CALIMA (full analysis set) and 31 from ZONDA.
The reduction in annual rate of asthma exacerbations observed in the placebo-controlled predecessor SIROCCO and CALIMA trials in patients with baseline blood eosinophil counts of ≥ 300 cells/microL who were taking high-dose ICS (ITT population) was maintained over the second year of treatment. In patients who received Fasenra in predecessor SIROCCO and CALIMA trials, 73% were exacerbation-free in the extension BORA trial. Similar maintenance of effect was observed throughout BORA in lung function, ACQ-6 and AQLQ(S)+12.
In a pre-specified pooled analysis, maintenance of the reduction in daily OCS dose was also observed over the extension trial in patients enrolled from ZONDA (Figure 2).
 MELTEMI was designed as an open-label safety extension study, enrolling adult patients who completed at least 16 weeks in ZONDA (see Section 4.8 Adverse Effects (Undesirable Effects)). The safety of the 2 dosing regimens studied in the predecessor studies was evaluated in 446 patients who received at least 1 dose of Fasenra.
MELTEMI was designed as an open-label safety extension study, enrolling adult patients who completed at least 16 weeks in ZONDA (see Section 4.8 Adverse Effects (Undesirable Effects)). The safety of the 2 dosing regimens studied in the predecessor studies was evaluated in 446 patients who received at least 1 dose of Fasenra.
Eosinophilic granulomatosis with polyangiitis (EGPA).
MANDARA. The efficacy and safety of Fasenra was evaluated in a randomised, double-blind, active-controlled, non-inferiority clinical trial of 52-weeks treatment duration, in patients aged 18 years and older with EGPA. A total of 140 patients who has a history of relapsing or refractory disease were randomised. Fasenra 30 mg was evaluated compared to mepolizumab 300 mg administered subcutaneously every 4 weeks in addition to background prednisolone/prednisone with or without immunosuppressive therapy. The OCS dose was tapered at the discretion of the investigator.
The demographics and baseline characteristics are provided in Table 8.

Remission.
The primary endpoint was the proportion of patients in remission, defined as Birmingham Vasculitis Activity Score (BVAS)=0 (no active vasculitis) plus prednisolone/prednisone dose ≤ 4 mg/day, at both Week 36 and Week 48. As shown in Table 9, Fasenra demonstrated non-inferiority to mepolizumab for the primary endpoint. Results for accrued duration of remission and the components of remission are also shown in Table 9.
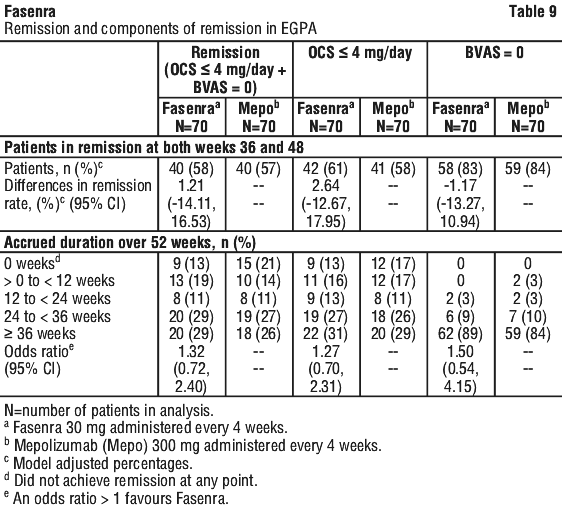 The proportion of patients achieving remission within the first 24 weeks of treatment and remaining in remission through Week 52 was 42% for Fasenra and 37% for mepolizumab (difference in responder rate 5.54%, 95% CI: -9.30, 20.37).
The proportion of patients achieving remission within the first 24 weeks of treatment and remaining in remission through Week 52 was 42% for Fasenra and 37% for mepolizumab (difference in responder rate 5.54%, 95% CI: -9.30, 20.37).
Using an alternative remission definition of BVAS=0 plus prednisolone/prednisone ≤ 7.5 mg/day, a consistent efficacy between groups for these endpoints was observed.
Patients achieved the primary remission endpoint across the prespecified demographic and baseline characteristic subgroups.
Relapse.
The hazard ratio for time to first relapse (defined as worsening related to vasculitis, asthma, or sino-nasal symptoms requiring an increase in dose of corticosteroids or immunosuppressive therapy or hospitalisation) was 0.98 (95% CI: 0.53, 1.82). Relapse was observed in 30% of patients on Fasenra and 30% of patients on mepolizumab. The annualised relapse rate was 0.50 for patients receiving Fasenra versus 0.49 for patients receiving mepolizumab (rate ratio 1.03, 95% CI: 0.56, 1.90). The types of relapse were consistent for patients receiving Fasenra or mepolizumab.
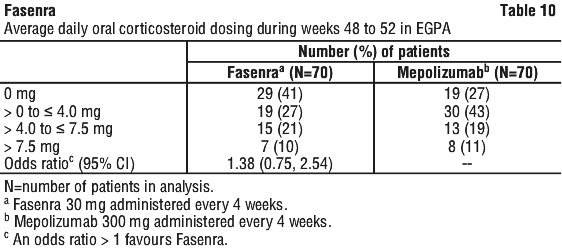
Oral corticosteroid reduction.
The average daily OCS dose during Weeks 48 to 52 is presented in Table 10. A 100% reduction in the OCS dose was observed in 41% of patients receiving Fasenra compared to 26% of those receiving mepolizumab (difference 15.69%, 95% CI: 0.67, 30.71). Reductions of 50% or higher were observed in 85% of patients receiving Fasenra compared to 74% of those receiving mepolizumab (difference 10.79%, 95% CI: -2.25, 23.83).
Asthma control questionnaire-6 (ACQ-6).
The ACQ-6 responder rate during Weeks 48 to 52 (defined as a decrease in score of 0.5 or more compared with baseline) was 42% for Fasenra and 48% for mepolizumab (difference -6.16%, 95% CI: -18.52, 6.21).
5.2 Pharmacokinetic Properties
The pharmacokinetic properties of benralizumab below are based on the population pharmacokinetic analyses from the asthma trials in adults and adolescents. Findings in EGPA were consistent with those in asthma. The pharmacokinetics of benralizumab were dose-proportional in patients with asthma following subcutaneous administration over a dose range of 2 to 200 mg.
Absorption.
Following subcutaneous administration to patients with asthma, the absorption half-life was 3.5 days. Based on population pharmacokinetic analysis, the estimated absolute bioavailability was approximately 59% and there was no clinically relevant difference in relative bioavailability in the administration to the abdomen, thigh or upper arm.
Distribution.
Based on population pharmacokinetic analysis, central and peripheral volume of distribution of benralizumab was 3.1 L and 2.5 L respectively for a 70 kg individual.
Metabolism.
Benralizumab is a humanized IgG1 monoclonal antibody that is degraded by proteolytic enzymes widely distributed in the body and not restricted to hepatic tissue.
Excretion.
From population pharmacokinetic analysis, benralizumab exhibited linear pharmacokinetics and no evidence of target receptor-mediated clearance pathway. The estimated systemic clearance (CL) for benralizumab was at 0.29 L/day. Following subcutaneous administration, the elimination half-life was approximately 15.5 days.
Special patient populations.
Elderly (≥ 65 years old).
Based on population pharmacokinetic analysis, age did not affect benralizumab clearance.
Gender, race.
A population pharmacokinetics analysis indicated that there was no significant effect of gender and race on benralizumab clearance.
Renal impairment.
No formal clinical studies have been conducted to investigate the effect of renal impairment on benralizumab. Based on population pharmacokinetic analysis, mild to moderate renal impairment (eGFR 30-89 mL/min/1.73 m2) did not affect benralizumab clearance. There are limited data available in patients with eGFR less than 30 mL/min/1.73 m2, however benralizumab is not cleared renally.
Hepatic impairment.
No formal clinical studies have been conducted to investigate the effect of hepatic impairment on benralizumab. IgG monoclonal antibodies are not primarily cleared via hepatic pathway; change in hepatic function is not expected to influence benralizumab clearance. Based on population pharmacokinetic analysis, baseline hepatic function biomarkers (ALT, AST and bilirubin) had no clinically relevant effect on benralizumab clearance.
Paediatric use.
Based on the population pharmacokinetic analysis, the pharmacokinetics of benralizumab in adolescents aged 12 to 17 years was consistent with adults. Benralizumab has not been studied in children below 12 years of age (see Section 4.2 Dose and Method of Administration, Special patient populations, Paediatric use).
5.3 Preclinical Safety Data
Genotoxicity.
No genotoxicity studies have been conducted. As a monoclonal antibody, benralizumab is not expected to interact with DNA or other chromosomal material.
Carcinogenicity.
Long-term animal studies have not been performed to evaluate the carcinogenic potential of benralizumab. Published literature using animal models suggests that IL-5 and eosinophils are part of an early inflammatory reaction at the site of tumorigenesis and can promote tumour rejection. However, other reports indicate that eosinophil infiltration into tumours can promote tumour growth. Therefore, the malignancy risk in humans from an antibody that binds to IL-5Rα, such as benralizumab, is unknown.6 Pharmaceutical Particulars
6.1 List of Excipients
Fasenra prefilled syringe and Fasenra Pen contain the excipients histidine, histidine hydrochloride monohydrate, trehalose, polysorbate 20 and water for injections.
Fasenra prefilled syringe and Fasenra Pen do not contain latex, lactose, sucrose, gluten, tartrazine or any other azo dyes.
6.2 Incompatibilities
Incompatibilities were not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store at 2°C to 8°C (Refrigerate. Do not freeze).
Fasenra may be kept at room temperature up to 25°C for a maximum of 14 days. After removal from the refrigerator, Fasenra must be used within 14 days or discarded.
Store Fasenra in the original package in order to protect from light. Do not freeze. Do not expose to heat.
6.5 Nature and Contents of Container
Fasenra prefilled syringe*.
Each prefilled syringe contains 30 mg benralizumab in 1 mL (30 mg/mL). The prefilled syringe is comprised of a type I glass barrel with a staked 29 gauge 12.7 mm stainless steel needle, rigid needle shield and FluoroTec-coated plunger stopper in a passive safety device to prevent needle stick injuries.
Fasenra prefilled syringe is available in a pack containing one single-dose, single use, sterile prefilled syringe.
Fasenra Pen (prefilled pen).
Each Fasenra Pen contains 30 mg benralizumab in 1 mL (30 mg/mL). The prefilled pen is comprised of an autoinjector made from type I glass with a staked 29 gauge 12.7 mm stainless steel needle, rigid needle shield, and Fluorotec-coated stopper in an autoinjector.
Fasenra Pen is available in a pack containing one single-dose, single-use, sterile prefilled pen.
*Not all presentations may be available in Australia.
6.6 Special Precautions for Disposal
Discard used Fasenra prefilled syringes/Fasenra Pen into a sharps disposal container.
In Australia, any unused medicine should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Benralizumab is a humanised, afucosylated, monoclonal antibody selective for the alpha subunit of the human interleukin-5 receptor (IL-5Rα). Benralizumab is of the IgG1, kappa-class produced in Chinese hamster ovary (CHO) cells by recombinant DNA technology. Benralizumab has a molecular weight of approximately 150 kDa.
Chemical structure.
General structure of benralizumab.

CAS number.
1044511-01-4.7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Summary Table of Changes
