
What is in this leaflet
This leaflet answers some common questions about Femara.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine.
You can also download the most up to date leaflet from www.novartis.com.au.
Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Femara against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Femara is used for
Femara is used to treat breast cancer in women who are post-menopausal - that is, women who no longer have periods, either naturally due to their age or after surgery or chemotherapy.
Femara is available in tablets containing 2.5 mg of the active ingredient, letrozole.
Letrozole belongs to a family of medicines called aromatase inhibitors. They are also called "antioestrogens" because they act by reducing the production of oestrogen in your body.
Oestrogen stimulates the growth of certain types of breast cancer. These cancers are called "oestrogen-dependent." Reducing the production of oestrogen may help to keep the cancer from growing.
This may be the first time you are taking an "antioestrogen" such as Femara or you may have taken another "antioestrogen" such as tamoxifen in the past.
Ask your doctor if you have any questions about why Femara has been prescribed for you. Your doctor may have prescribed Femara for another reason.
Femara is only available with a doctor's prescription. It is not addictive.
Before you take Femara
When you must not use it
Do not take Femara if you have an allergy to:
- letrozole, the active ingredient in Femara
- any other ingredients of Femara listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include rash, itching or hives on the skin; swelling of the face, lips, tongue or other parts of the body; shortness of breath, wheezing or troubled breathing.
Do not take Femara if you are still having periods. This medicine is only used in women who are no longer having periods.
If you recently became postmenopausal or if you are perimenopausal, you should discuss with your doctor about the necessity of contraception as you might have the potential to become pregnant.
Do not take Femara if you are pregnant. It may harm your unborn baby if you take it while you are pregnant. Your doctor will discuss with you the potential risks of taking Femara during pregnancy. There are reports of abnormalities in babies born to mothers who took Femara during pregnancy.
Do not take Femara if you are breast feeding.
Do not take Femara after the use by (expiry) date printed on the pack. If you take this medicine after the expiry date has passed, it may not work or it may make you unwell.
Do not take Femara if the packaging is torn or shows signs of tampering. In that case, return it to your pharmacist.
Before you start to take it
Tell your doctor if you have severe kidney or liver disease. Your doctor may want to take special precautions while you are taking this medicine.
Tell your doctor if you have a history of osteoporosis or bone fractures.
Your level of hormones may be checked by your doctor before you take Femara to ensure you have gone through the menopause (cessation of periods).
Tell your doctor if you are allergic to any other medicines, foods, dyes or preservatives. Your doctor will want to know if you are prone to allergies.
If you have not told your doctor about any of these things, tell them before you take Femara.
Taking other medicines
Tell your doctor if you are taking any other medicines, including medicines that you buy without a prescription from a pharmacy, supermarket or health food shop. Other medicines may be affected by Femara or they may affect how well it works.
Your doctor or pharmacist can tell you what to do when taking Femara with other medicines. This includes in particular:
- tamoxifen.
- other anti-estrogens or estrogen-containing therapies.
These substances may diminish the action of Femara.
Females of child-bearing potential and male patients
If you still until recently had menstrual periods, you should discuss with your doctor about the necessity of effective contraception as you might have the potential to become pregnant. Ask your doctor about options of effective birth control.
Femara may reduce fertility in male patients.
How to take Femara
Follow the directions given to you by your doctor and pharmacist carefully. These directions may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
The usual dose is one Femara tablet daily.
How to take it
Swallow the tablet with a glass of water or other liquid.
If your stomach is upset after taking the tablet, take it with a meal or after a snack.
How long to take it
Your doctor will check your progress to make sure the medicine is working and will decide how long your treatment should continue.
If you are unsure, talk to your doctor.
If you forget to take it
If it is almost time for your next dose (e.g. within 2 or 3 hours), skip the dose you missed and take your next dose when you are meant to.
Otherwise, take the dose as soon as you remember, and then go back to taking your tablet as you would normally.
Do not take a double dose to make up for the one that you missed.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much
Immediately telephone your doctor or Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at your nearest hospital if you think that you or anyone else may have taken too much Femara. Do this even if there are no signs of discomfort or poisoning. Keep the telephone numbers for these places handy.
While you are taking Femara
Things you must do
If you become pregnant while taking Femara, tell your doctor immediately. You should not take this medicine while you are pregnant.
Follow your doctor's instructions carefully. If you do not follow your doctor's instructions, your treatment may not help or you may have unwanted side effects.
Be sure to keep all of your doctor's appointments so that your progress can be checked. Your doctor may want you to have blood tests from time to time to check on your progress and detect any unwanted side effects. Your doctor may also decide to monitor your bone health as this medicine may cause thinning or wasting of your bones (osteoporosis).
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Femara.
Tell any other doctor, dentist or pharmacist who treats you that you are taking Femara.
Things you must not do
Do not use Femara to treat any other complaints unless your doctor says you can.
Do not give this medicine to anyone else, even if their symptoms seem to be similar to yours.
Things to be careful of
Be careful driving, operating machinery or doing jobs that require you to be alert while you are taking Femara until you know how it affects you.
This medicine may cause dizziness or tiredness in some people. If you have any of these symptoms, do not drive or do anything else that could be dangerous.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Femara.
It may have unwanted side effects in some people in addition to its beneficial effects. All medicines have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- signs that blood clots may have formed, such as sudden severe headache, sudden loss of coordination, blurred vision or sudden loss of vision, slurred speech, numbness or tingling in an arm or leg, painful swelling in the calves or thighs, chest pain, difficulty breathing, coughing blood, rapid heartbeat, bluish skin discolouration, fainting.
- constant "flu-like" symptoms (chills, fever, sore throat, sores in mouth, swollen glands, tiredness or lack of energy) that could be a sign of blood problems.
- swelling mainly of the face and throat (signs of allergic reaction)
- weakness or paralysis of limbs or face, difficulty speaking (signs of stroke)
- crushing chest pain or sudden arm or leg (foot) pain (signs of a heart attack)
- swelling and redness along a vein which is extremely tender, possibly painful to touch (signs of thrombophlebitis)
The above side effects may be serious. You may need urgent medical attention or hospitalisation.
Tell your doctor straight away if you experience any of the following:
- yellow skin and eyes, nausea, loss of appetite, dark coloured urine (signs of hepatitis)
- rash, red skin, blistering of the lips, eyes or mouth, skin peeling, fever (signs of skin disorder)
- blurred vision (sign of cataract)
- swelling of the feet, ankles or other parts of the body due to fluid build-up (signs of oedema)
Tell your doctor if you notice any of the following side effects and they worry you:
- skin rash, itching or dry skin
- pain in the muscles, joints or bones; joint stiffness, arthritis, back pain
- high level of cholesterol
- vaginal spotting or bleeding
- whitish, thick vaginal discharge, vaginal dryness
- headache
- fever
- tiredness, sleepiness, weakness or dizziness, vertigo
- fall
- chest pain
- difficulty sleeping
- numbness or tingling in hands or feet
- mood changes such as anxiety, nervousness, irritability and depression (sad mood)
- drowsiness
- forgetfulness
- blurred vision or eye irritation
- stomach upset, nausea (feeling sick) or vomiting, indigestion, pain in the abdomen
- constipation
- diarrhoea
- dry mouth, sore mouth, mouth ulcers and cold sores
- thirst, change in sense of taste, dry mouth
- dry mucous membranes of the mouth, nose, vagina
- breast pain
- hot flushes
- increased sweating
- appetite changes
- increase or decrease in weight
- hair thinning
- urgent need to urinate (pass water)
- pain or burning sensation when urinating, which may be a sign of an infection
- pain or burning sensation in the hands or wrist (carpal tunnel syndrome)
- fast or irregular heartbeat, palpitations, high blood pressure (hypertension)
- thinning of bones (osteoporosis), bone fractures
- cough
- trigger finger, a condition in which your finger or thumb catches in a bent position
- dark coloured urine
- yellowish eyes and/or skin (jaundice)
Tell your doctor if you notice anything else that is making you unwell.
Other side effects not listed above may happen in some people.
Some of these can only be found by laboratory testing.
After taking Femara
Storage
- Keep your tablets in the container until it is time to take them.
- Store the tablets in a cool dry place below 30°C (room temperature).
- Do not store Femara in the bathroom or any other place that is hot or steamy.
- Do not leave the tablets in the car or on window sills.
Heat and dampness can destroy some medicines. Femara will keep well if it is cool and dry.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking Femara or the tablets have passed their use by (expiry date), ask your pharmacist what to do with any that are left over.
Product description
What it looks like
Femara 2.5 mg: dark yellow, round, coated tablet, marked CG on one side and FV on the other; supplied in blister packs in a cardboard carton of 30 tablets.
Ingredients
Femara tablets contain 2.5 mg of letrozole as the active ingredient.
The tablets also contain:
- silica - colloidal anhydrous
- cellulose - microcrystalline
- lactose
- magnesium stearate
- starch-maize
- sodium starch glycollate
- hypromellose
- iron oxide yellow
- macrogol 8000
- talc - purified
- titanium dioxide
Femara does not contain sucrose, gluten, tartrazine or any other azo dyes. Femara contains lactose, galactose, milk, sulfites, sugars and ethanol.
Sponsor
Femara is supplied in Australia by:
NOVARTIS Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
Macquarie Park NSW 2113
Telephone 1-800-671-203
Australian Registration Number
AUST R 60605
Date of preparation
This leaflet was prepared in May 2020
® = Registered trademark
(fem270520c.doc based on PI fem270520i.doc)
Published by MIMS July 2020



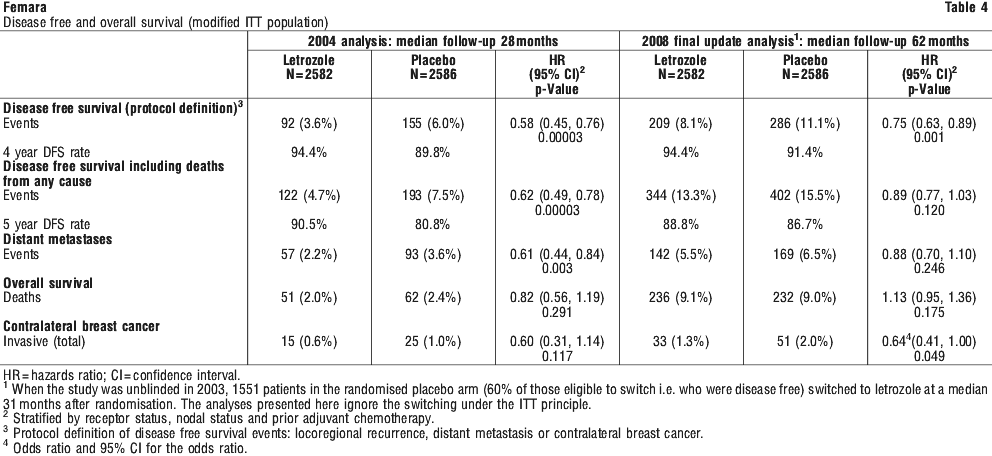
 In the updated analysis, as shown in Table 4, there was a significant reduction in the odds of an invasive contralateral breast cancer with Femara compared with placebo, despite 60% of the patients in the placebo arm having switched to Femara. There was no significant difference in overall survival.
In the updated analysis, as shown in Table 4, there was a significant reduction in the odds of an invasive contralateral breast cancer with Femara compared with placebo, despite 60% of the patients in the placebo arm having switched to Femara. There was no significant difference in overall survival. Both time to progression and objective response rate were significantly longer/ higher for letrozole than for tamoxifen irrespective of receptor status (see Table 7).
Both time to progression and objective response rate were significantly longer/ higher for letrozole than for tamoxifen irrespective of receptor status (see Table 7). Study design allowed patients to cross over upon progression to the other therapy or discontinue from the study. Approximately 50% of patients crossed over to the opposite treatment arm and crossover was virtually completed by 36 months. The median time to crossover was 17 months (Femara to tamoxifen) and 13 months (tamoxifen to Femara). Femara treatment in the first line therapy of advanced breast cancer patients is associated with an early survival advantage over tamoxifen. The median survival was 34 months for Femara and 30 months for tamoxifen. A significantly greater number of patients were alive on Femara versus tamoxifen throughout the first 24 months of the study (repeated log rank test) (see Table 8).
Study design allowed patients to cross over upon progression to the other therapy or discontinue from the study. Approximately 50% of patients crossed over to the opposite treatment arm and crossover was virtually completed by 36 months. The median time to crossover was 17 months (Femara to tamoxifen) and 13 months (tamoxifen to Femara). Femara treatment in the first line therapy of advanced breast cancer patients is associated with an early survival advantage over tamoxifen. The median survival was 34 months for Femara and 30 months for tamoxifen. A significantly greater number of patients were alive on Femara versus tamoxifen throughout the first 24 months of the study (repeated log rank test) (see Table 8). In patients who did not cross over to the opposite treatment arm, median survival was 35 months with Femara (N = 219, 95% CI 29 to 43 months) vs. 20 months with tamoxifen (N = 229, 95% CI 16 to 26 months).
In patients who did not cross over to the opposite treatment arm, median survival was 35 months with Femara (N = 219, 95% CI 29 to 43 months) vs. 20 months with tamoxifen (N = 229, 95% CI 16 to 26 months).
