1 Name of Medicine
Fluconazole.
2 Qualitative and Quantitative Composition
Fluconazole is a member of the bis-triazole class of antifungal agents.
Fluconazole is a white crystalline powder, slightly soluble in water, freely soluble in methanol.
Fluconazole-WGR (fluconazole) capsules contain 50, 100 and 200 mg of the active ingredient, fluconazole.
Excipient with known effects.
Contains sugars (as lactose). For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Fluconazole-WGR is available as:
Blue/ white hard gelatin, self locked capsules of size '4' imprinted with 'RANBAXY' in black edible ink on both cap and body containing white to off white powder, containing 50 mg of fluconazole.
Blue/ white hard gelatin, self locked capsules of size '2' imprinted with 'RANBAXY' in black edible ink on both cap and body containing white to off white powder, containing 100 mg of fluconazole.
Purple/ white hard gelatin, self locked capsules of size '0' imprinted with 'RANBAXY' in black edible ink on both cap and body containing white to off white powder, containing 200 mg of fluconazole.
4.1 Therapeutic Indications
Fluconazole-WGR (fluconazole) capsules, given orally, are indicated for the following conditions:
Treatment of cryptococcal meningitis in patients who are unable to tolerate amphotericin B.
Note.
Data suggest that the clinical efficacy of fluconazole is lower than that of amphotericin B in the treatment of the acute phase of cryptococcal meningitis.
Maintenance therapy to prevent relapse of cryptococcal meningitis in patients with AIDS.
Treatment of oropharyngeal and oesophageal candidiasis in AIDS and other immunosuppressed patients.
Secondary prophylaxis of oropharyngeal candidiasis in patients with HIV infection.
Serious and life-threatening Candida infections in patients who are unable to tolerate amphotericin B.
Note.
It remains to be shown that fluconazole is as effective as amphotericin B in the treatment of serious and life-threatening Candida infections. Until such data are available, amphotericin B remains the drug of choice.
Vaginal candidiasis, when topical therapy has failed.
Treatment of extensive tinea corporis, extensive tinea cruris and extensive tinea pedis infections in immunocompetent patients in whom topical therapy is not a practical treatment option. Usually, topical therapy should be attempted first because oral therapy has a less favourable ratio of benefits to risks (see Section 4.8 Adverse Effects (Undesirable Effects)).4.2 Dose and Method of Administration
Method of administration.
Fluconazole-WGR capsules are intended for oral administration.
Dosage.
Adults. For cryptococcal meningitis in patients who are unable to take or tolerate amphotericin B.
The usual dose is 400 mg on the first day followed by 200 mg once daily. A dosage of 400 mg once daily may be used, based on medical judgement of the patient's response to therapy. Patients not responding to treatment for up to 60 days would appear unlikely to respond to fluconazole.
Duration of treatment for cryptococcal infections will depend on the clinical and mycological response, but should continue 10-12 weeks after cerebrospinal fluid becomes culture negative. Negative serology does not necessarily indicate eradication of the disease; a proportion of such patients relapse in due course.
Prevention of relapse of cryptococcal meningitis in AIDS patients.
After the patient receives a full course of primary therapy, fluconazole may be administered at a daily dose of 100-200 mg.
Oropharyngeal candidiasis.
The recommended dose is 100 mg on the first day followed by 50 mg once daily. For the treatment of oesophageal candidiasis, the recommended dose is 200 mg on the first day, followed by 100 mg once daily. Clinical evidence of candidiasis usually resolves within several days, but treatment should be continued for at least two to three weeks especially in patients with severely compromised immune function. Patients with severe oesophageal candidiasis may need treatment to be continued for two weeks following resolution of symptoms. Approximately half of the clinically cured patients remain colonised.
Secondary prophylaxis against oropharyngeal candidiasis in patients with HIV infections.
The recommended dose is 150 mg as a single dose once weekly.
Serious and life-threatening candidal infections in patients unable to tolerate amphotericin B.
The usual dose is 400 mg on the first day followed by 200 mg daily. Depending on the clinical response, the dose may be increased to 400 mg daily. Duration of treatment is based on clinical response; patients should be treated for minimum of 4 weeks and for at least 2 weeks following resolution of symptoms.
Vaginal candidiasis when topical therapy has failed.
Fluconazole 150 mg should be administered as a single oral dose.
In those patients who responded to treatment, the median time to onset of symptom relief was one day (range: 0.04-9 days) and to complete symptom relief was two days (range: 0.5-20 days).
For extensive tinea infections (tinea corporis, tinea cruris), or severe tinea pedis in immunocompetent patients in whom topical therapy is not practical.
The recommended dosage is 150 mg once weekly for 4 weeks.
Children. Fluconazole capsules are not suitable for children weighing under 35 kg.
As with similar infections in adults, the duration of treatment is based on the clinical and mycological response. Fluconazole is administered as a single dose each day.
Mucosal candidiasis.
The recommended dosage is 3 mg/kg daily. A loading dose of 6 mg/kg may be used on the first day to achieve steady-state levels more rapidly.
Systemic candidiasis and cryptococcal infection.
The recommended dosage is 6 to 12 mg/kg daily, depending on the severity of the disease.
For children with impaired renal function the daily dose should be reduced in accordance with the guidelines given for adults.
Elderly. Dose should be adjusted for elderly patients with renal impairment (see Renal impairment).
Renal impairment. Fluconazole is predominantly excreted in the urine as unchanged drug. No adjustments in single-dose therapy are necessary. In multiple-dose treatment of patients with renal impairment, normal doses should be given on days 1 and 2 of treatment and thereafter the dosage intervals or the daily dose should be modified in accordance with creatinine clearances as follows (see Table 1):

Patients receiving regular dialysis.
One recommended dose after every dialysis session. These are suggested dose adjustments based on pharmacokinetics following administration of single doses. Further adjustment may be needed depending on clinical condition.
When serum creatinine is the only measure of renal function available, the following formula (based on sex, weight, and age of patient) should be used to estimate the creatinine clearance in mL/minute (see Equation 1).

4.3 Contraindications
Fluconazole-WGR (fluconazole) capsules are contraindicated in the following conditions:
Patients with known sensitivity to fluconazole; to related azole compounds; or to any of its excipients.
Concomitant administration with cisapride.
Co-administration of terfenadine is contraindicated in patients receiving fluconazole at multiple doses of 400 mg/day or higher based upon results of a multiple dose interaction study.
Co-administration of other drugs known to prolong the QT interval and which are metabolised via the enzyme CYP3A4 such as cisapride, astemizole, erythromycin, pimozide and quinidine are contraindicated (see Section 4.4 Special Warnings and Precautions for Use).
4.4 Special Warnings and Precautions for Use
General.
In rare cases, as with other azoles, anaphylaxis has been reported. Patients have rarely developed exfoliative cutaneous reactions, such as Stevens-Johnson syndrome and toxic epidermal necrolysis, during treatment with fluconazole. Drug reaction with eosinophilia and systemic symptoms (DRESS) has been reported. AIDS patients are more prone to the development of serious cutaneous reactions to many drugs. If rash which is attributable to fluconazole develops in a patient treated for a superficial fungal infection, fluconazole should be discontinued. If patients with invasive/systemic fungal infections develop rashes, they should be monitored closely and fluconazole discontinued if bullous lesions or erythema multiforme develop (see Section 4.8 Adverse Effects (Undesirable Effects)).
Some azoles, including fluconazole, have been associated with prolongation of the QT interval on the electrocardiogram. Fluconazole causes QT prolongation via the inhibition of Rectifier Potassium Channel current. The QT prolongation caused by other medicinal products (such as amiodarone) may be amplified via the inhibition of cytochrome P450 (CYP3A4) (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). During post-marketing surveillance, there have been very rare cases of QT prolongation and torsades de pointes in patients taking fluconazole. These reports included seriously ill patients with multiple confounding risk factors, such as structural heart disease, electrolyte abnormalities and concomitant medications that may have been contributory. Patients with hypokalaemia and advanced cardiac failure are at an increased risk for the occurrence of life-threatening ventricular arrhythmias and torsades de pointes. Fluconazole should be administered with caution to patients with these potentially proarrhythmic conditions (see Section 4.8 Adverse Effects (Undesirable Effects)).
Adrenal insufficiency has been reported in patients receiving other azoles (e.g. ketoconazole).
Cases of adrenal insufficiency were reported in patients receiving fluconazole.
Fluconazole is a potent CYP2C9 and CYP2C19 inhibitor and a moderate CYP3A4 inhibitor. Fluconazole-treated patients who are concomitantly treated with drugs with a narrow therapeutic window metabolized through CYP2C9, CYP2C19 and CYP3A4 should be monitored (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Fluconazole capsules contain lactose monohydrate and should not be given to patients with rare hereditary problems of galactose intolerance, Lapp-lactase deficiency or glucose-galactose malabsorption.
Candidiasis.
Studies have shown an increasing prevalence of infections with Candida species other than C. albicans. These are often resistant (e.g. C. krusei and C. auris) or show reduced susceptibility to fluconazole (C. glabrata). Such infections may require alternative antifungal therapy secondary to treatment failure. Therefore, prescribers are advised to take into account the prevalence of resistance in various Candida species to fluconazole (see Section 5.1 Pharmacodynamic Properties).
Use in hepatic impairment.
Fluconazole should be administered with caution to patients with liver dysfunction.
Fluconazole has been associated with rare cases of serious hepatic toxicity including fatalities, primarily in patients with serious underlying medical conditions. In cases of fluconazole-associated hepatotoxicity, no obvious relationship to total daily dose, duration of therapy, sex or age of patient has been observed.
Patients who develop abnormal liver function tests during fluconazole therapy should be monitored for the development of more severe hepatic injury. Fluconazole should be discontinued if clinical signs and symptoms consistent with liver disease develop that may be attributable to fluconazole.
Use in renal impairment.
Fluconazole should be administered with caution to patients with renal dysfunction.
Use in the elderly.
Dosage should be adjusted for elderly patients with renal impairment (see Section 4.2 Dose and Method of Administration).
Pediatric use.
See Section 4.2 Dose and Method of Administration, Dosage, Children; Section 5.2 Pharmacokinetic Properties, Children.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Fluconazole is an inhibitor of the cytochrome P450 system, particularly the CYP 2C and to a lesser extent the CYP 3A isoforms. Co-administration of fluconazole with some other drugs metabolized primarily by these P450 isoforms may result in altered plasma concentrations of these drugs that could change therapeutic effects and/or adverse event profiles.
Clinically or potential significant drug interactions have been observed between fluconazole and the following agents: short acting benzodiazepines, cisapride, coumarin-type anticoagulants, ciclosporin, hydrochlorothiazide, oral hypoglycaemics, phenytoin, rifampicin, rifabutin, tacrolimus and theophylline. These are described in greater detail below.
Effect of other medicinal products on fluconazole.
The exposure to fluconazole is significantly increased by the concomitant administration of the following agent. Hydrochlorothiazide.
Concomitant oral administration of 100 mg fluconazole and 50 mg hydrochlorothiazide for 10 days in normal volunteers resulted in an increase of 41% in Cmax and an increase of 43% in AUC of fluconazole, compared to fluconazole given alone. Overall, the plasma concentrations of fluconazole were approximately 3.26 - 6.52 micromol/L higher with concomitant diuretic. These changes are attributable to a mean net reduction of approximately 20% in renal clearance of fluconazole.
The exposure to fluconazole is significantly decreased by the concomitant administration of the following agent. Rifampicin.
Administration of a single oral 200 mg dose of fluconazole after chronic rifampicin administration resulted in a 25% decrease in AUC and a 20% shorter half-life of fluconazole in normal volunteers. Depending on clinical circumstances, an increase of the dose of fluconazole should be considered when it is administered with rifampicin.
Minor or no significant pharmacokinetic interactions that require no dosage adjustment. Gastrointestinal drugs.
In fasted normal volunteers, absorption of orally administered fluconazole does not appear to be affected by agents that increase gastric pH. Single dose administration of fluconazole (100 mg) with cimetidine (400 mg) resulted in a 13% reduction in AUC and a 21% reduction in Cmax of fluconazole. Administration of an antacid containing aluminium and magnesium hydroxides immediately prior to a single dose of fluconazole (100 mg) had no effect on the absorption or elimination of fluconazole.
Effects of fluconazole on other medicinal products.
Fluconazole is a potent inhibitor of cytochrome P450 (CYP) isoenzymes 2C9 and 2C19 and a moderate inhibitor of CYP3A4. In addition to the observed /documented interactions mentioned below, there is a risk of increased plasma concentration of other compounds metabolised by CYP2C9, CYP2C19 and CYP3A4 co-administered with fluconazole. Therefore, caution should be exercised when using these combinations and the patients should be carefully monitored. The enzyme inhibiting effect of fluconazole persists 4 to 5 days after discontinuation of fluconazole treatment due to the long half-life of fluconazole (see Section 4.3 Contraindications).
Abrocitinib.
Fluconazole (inhibitor of CYP2C19, 2C9, 3A4) increased exposure of abrocitinib active moiety by 155%. If coadministered with fluconazole, adjust the dose of abrocitinib as instructed in the abrocitinib product information.
Alfentanil.
A study observed a reduction in clearance and distribution volume as well as prolongation of t1/2 of alfentanil following concomitant treatment with fluconazole. A possible mechanism of action is fluconazole's inhibition of CYP3A4. Dosage adjustment of alfentanil may be necessary.
Amitriptyline, nortriptyline.
Fluconazole increases the effect of amitriptyline and nortriptyline. 5-nortriptyline and/or S-amitriptyline may be measured at initiation of the combination therapy and after 1 week. Dosage of amitriptyline/nortriptyline should be adjusted, if necessary.
Amphotericin B.
Concurrent administration of fluconazole and amphotericin B in infected normal and immunosuppressed mice showed the following results: a small additive antifungal effect in systemic infection with Candida albicans, no interaction in intracranial infection with Cryptococcus neoformans, and antagonism of the two drugs in systemic infection with Aspergillus fumigatus. The clinical significance of results obtained in these studies is unknown.
Concomitant use of the following agents with fluconazole is contraindicated. Cisapride.
Fluconazole 200 mg daily increased the AUC and Cmax of cisapride (20 mg four times daily) both after a single dose (AUC increased 101% and Cmax increased 91%) and multiple doses (AUC increased 192% and Cmax increased 154%). A significant prolongation in QTc interval was recorded. Cardiac events including torsades de pointes have been reported in patients receiving fluconazole and cisapride concomitantly. In most of these cases the patients appear to have been predisposed to arrhythmias or had serious underlying illness. The co-administration of fluconazole and cisapride is contraindicated.
Astemizole.
Concomitant administration of fluconazole with astemizole may decrease the clearance of astemizole. Resulting increased plasma concentrations of astemizole can lead to QT prolongation and rare occurrences of torsades de pointes. Co-administration of fluconazole and astemizole is contraindicated (see Section 4.3 Contraindications).
Erythromycin.
Concomitant use of fluconazole and erythromycin has the potential to increase the risk of cardiotoxicity (prolonged QT interval, torsades de pointes) and consequently sudden heart death. Co-administration of fluconazole and erythromycin is contraindicated.
Pimozide.
Although not studied in vitro or in vivo, concomitant administration of fluconazole with pimozide may result in inhibition of pimozide metabolism. Increased pimozide plasma concentrations can lead to QT prolongation and rare occurrences of torsades de pointes. Co-administration of fluconazole and pimozide is contraindicated (see Section 4.3 Contraindications).
Quinidine.
Although not stated in vitro or in vivo, concomitant administration of fluconazole with quinidine may result in inhibition of quinidine metabolism. Use of quinidine has been associated with QT prolongation and rare occurrences of torsades de pointes. Co-administration of fluconazole and quinidine is contraindicated (see Section 4.3 Contraindications).
Terfenadine.
Because of the occurrence of serious cardiac dysrhythmias secondary to prolongation of the QTc interval in patients receiving azole antifungals in conjunction with terfenadine, interaction studies have been performed. One study at a 200 mg daily dose of fluconazole failed to demonstrate a prolongation in QTc interval. Another study at a 400 mg and 800 mg daily dose of fluconazole demonstrated that fluconazole taken in doses of 400 mg per day or greater significantly increases plasma levels of terfenadine when taken concomitantly. The combined use of fluconazole at doses of 400 mg or greater with terfenadine is contraindicated (see Section 4.3 Contraindications). The co-administration of fluconazole at doses lower than 400 mg per day with terfenadine should be carefully monitored.
Concomitant use that should be avoided or used with caution. Amiodarone.
Concomitant administration of fluconazole with amiodarone may increase QT prolongation. Caution must be exercised if the concomitant use of fluconazole and amiodarone is necessary, notably with high-dose fluconazole (800 mg).
Lemborexant.
Concomitant administration of fluconazole increased lemborexant Cmax and AUC by approximately 1.6- and 4.2-fold, respectively which is expected to increase risk of adverse reactions, such as somnolence. Avoid concomitant use of lemborexant.
Interaction of fluconazole with the following agents may result in increased exposure to these drugs. Careful monitoring and/or dosage adjustment should be considered. Anticoagulants.
Careful monitoring of prothrombin time in patients receiving fluconazole and indanedione anticoagulants is recommended.
Benzodiazepines (short acting).
Studies in human subjects have reported changes in midazolam pharmacokinetics and clinical effects that are dependent on dosage and route of administration. Single doses of fluconazole 150 mg resulted in modest increases in midazolam concentrations and psychomotor effects following oral administration of 10 mg that may not be clinically significant. At doses used to treat systemic mycoses, fluconazole resulted in substantial increases in midazolam concentrations and psychomotor effects following oral administration of 7.5 mg, but only modest increases that are not likely to be clinically significant following intravenous infusion of midazolam 0.05 mg/kg. This effect on midazolam appears to be more pronounced following oral administration of fluconazole than with fluconazole administered intravenously. There have been reports of sleepiness and disturbed consciousness in patients taking fluconazole for systemic mycoses and triazolam. However, in most of these cases the patients had serious underlying illnesses and/or concomitant therapies that could have contributed to the reported events, and a true fluconazole-triazolam interaction has not been established. If concomitant benzodiazepine therapy is necessary in patients being treated with fluconazole, consideration should be given to decreasing the benzodiazepine dosage, and the patients should be appropriately monitored. Fluconazole increases the AUC of triazolam (single dose) by approximately 50% Cmax with 20% to 32% and increases the half-life by 25% to 50% due to the inhibition of metabolism of triazolam. Dosage adjustments of triazolam may be necessary.
Carbamazepine.
Azole antifungals may raise carbamazepine plasma concentrations. Since high plasma concentrations of carbamazepine and/or cabamazepine-10, 11-epoxy may result in adverse effects (e.g. dizziness, drowsiness, ataxia, diplopia), the dosage of carbamazepine should be adjusted accordingly and/or plasma concentrations monitored when used concomitantly with fluconazole.
Calcium channel blockers.
Certain calcium channel antagonists (nifedipine, isradipine, amlodipine, verapamil and felodipine) are metabolised by CYP3A4. Fluconazole has the potential to increase the systemic exposure of the calcium channel antagonists. Frequent monitoring for adverse events is recommended.
Celecoxib.
During concomitant treatment with fluconazole (200 mg daily) and celecoxib (200 mg) the celecoxib Cmax and AUC increased by 68% and 134%, respectively. Half of the celecoxib dose may be necessary when combined with fluconazole.
Ciclosporin.
Fluconazole significantly increases the concentration and AUC of ciclosporin. This combination may be used by reducing the dosage of ciclosporin depending on ciclosporin concentration.
Cyclophosphamide.
Combination therapy with cyclophosphamide and fluconazole results in an increase in serum bilirubin and serum creatinine. The combination may be used while taking increased consideration to the risk of increased serum bilirubin and serum creatinine.
Everolimus.
Although not studied in vivo or in vitro, fluconazole may increase serum concentrations of everolimus through inhibition of CYP3A4.
Fentanyl.
One fatal case of possible fentanyl fluconazole interaction was reported. The author judged that the patient died from fentanyl intoxication. Furthermore, in a randomised crossover study with 12 healthy volunteers it was shown that fluconazole delayed the elimination of fentanyl significantly. Elevated fentanyl concentration may lead to respiratory depression.
Halofantrine.
Fluconazole can increase halofantrine plasma concentration due to an inhibitory effect on CYP3A4.
HMG-CoA reductase inhibitors.
The risk of myopathy and rhabdomyolysis increases (dose dependent) when fluconazole is co-administered with HMG-CoA reductase inhibitors metabolised through CYP3A4, such as atorvastatin and simvastatin, or through CYP2C9, such as fluvastatin (decreased hepatic metabolism of the statin). If concomitant therapy is necessary, the patient should be observed for symptoms of myopathy and rhabdomyolysis and creatine kinase should be monitored. HMG-CoA reductase inhibitors should be discontinued if a marked increase in creatine kinase is observed, or myopathy/rhabdomyolysis is diagnosed or suspected. Lower doses of HMG-CoA reductase inhibitors may be necessary as instructed in the statin prescribing information.
Ibrutinib.
Moderate inhibitors of CYP3A4 such as fluconazole increase plasma ibrutinib concentrations and may increase risk of toxicity. If the combination cannot be avoided, reduce the dose of ibrutinib as instructed in ibrutinib prescribing information and provide close clinical monitoring.
Ivacaftor (alone or combined with drugs in the same therapeutic class).
Coadministration with ivacaftor, a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator, increased ivacaftor exposure by 3-fold. A reduction of the ivacaftor (alone or combined) dose is necessary as instructed in the ivacaftor (alone or combined) prescribing information.
Losartan.
Fluconazole inhibits the metabolism of losartan to its active metabolite (E-31 74) which is responsible for most of the angiotensin II-receptor antagonism that occurs during treatment with losartan. Patients should have their blood pressure monitored continuously.
Lurasidone.
Moderate inhibitors of CYP3A4 such as fluconazole may increase lurasidone plasma concentrations. If concomitant use cannot be avoided, reduce the dose of lurasidone as instructed in the lurasidone prescribing information.
Methadone.
Fluconazole may enhance the serum concentration of methadone. Dosage adjustment of methadone may be necessary.
Non-steroidal anti-inflammatory drugs.
Although not specifically studied, fluconazole has the potential to increase the systemic exposure of other non-steroidal anti-inflammatory drugs (NSAIDs) that are metabolised by CYP2C9 (e.g. naproxen, lornoxicam, meloxicam, diclofenac). Frequent monitoring for adverse events and toxicity related to NSAIDs is recommended. Adjustment of dosage of NSAIDs may be needed.
Olaparib.
Moderate inhibitors of CYP3A4 such as fluconazole increase olaparib plasma concentrations; concomitant use is not recommended. If the combination cannot be avoided, reduce the dose of olaparib as instructed in the Lynparza (Olaparib) Prescribing Information.
Oral hypoglycaemic agents.
The effects of fluconazole on the pharmacokinetics of the sulphonylurea oral hypoglycaemic agents tolbutamide, glipizide and glibenclamide were examined in three placebo-controlled crossover studies in normal volunteers. All subjects received the sulphonylurea alone and following treatment with 100 mg of fluconazole as a single daily oral dose for 7 days. Fluconazole administration resulted in significant increases in Cmax and AUC of the sulphonylurea. Several subjects in these three studies experienced symptoms consistent with hypoglycaemia. In the glibenclamide study, several volunteers required oral glucose treatment. When fluconazole and sulphonylureas are co-administered, blood glucose concentrations should be monitored carefully and the dose of the sulphonylurea adjusted accordingly.
Phenytoin.
Fluconazole inhibits the hepatic metabolism of phenytoin. With co-administration, serum phenytoin concentration levels should be monitored in order to avoid phenytoin toxicity.
Prednisone.
There was a case report that a liver-transplanted patient treated with prednisone developed acute adrenal cortex insufficiency when a 3-month therapy with fluconazole was discontinued. The discontinuation of fluconazole presumably caused an enhanced CYP3A4 activity which led to increased metabolism of prednisone. Patients on long-term treatment with fluconazole and prednisone should be carefully monitored for adrenal cortex insufficiency when fluconazole is discontinued.
Rifabutin.
There have been reports that an interaction exists when fluconazole is administered concomitantly with rifabutin, leading to increased serum levels of rifabutin up to 80%. There have been reports of uveitis in patients to whom fluconazole and rifabutin were co-administered. Patients receiving rifabutin and fluconazole concomitantly should be carefully monitored.
Saquinavir.
Fluconazole increases the AUC of saquinavir and decreases the clearance of saquinavir due to inhibition of saquinavir's hepatic metabolism by CYP3A4 and inhibition of P-glycoprotein. Dosage adjustment of saquinavir may be necessary.
Sirolimus.
Fluconazole increases plasma concentrations of sirolimus presumably by inhibiting the metabolism of sirolimus via CYP3A4 and P-glycoprotein. This combination may be used with a dosage adjustment of sirolimus depending on the effect/concentration measurements.
Sulfonylureas.
Fluconazole has been shown to prolong the serum half-life of concomitantly administered oral sulfonylureas (e.g. chlorpropamide, glibenclamide, glipizide, tolbutamide) in healthy volunteers. Frequent monitoring of blood glucose and appropriate reduction of sulfonylurea dosage are recommended during co-administration.
Tacrolimus.
Fluconazole may increase the serum concentrations of orally administered tacrolimus up to 5 times due to inhibition of tacrolimus metabolism through CYP3A4 in the intestines. No significant pharmacokinetic changes have been observed when tacrolimus is given intravenously. Increased tacrolimus levels have been associated with nephrotoxicity. Dosage of orally administered tacrolimus should be decreased depending on tacrolimus concentration.
Theophylline.
In a placebo-controlled interaction study, the administration of fluconazole 200 mg for 14 days resulted in an 18% decrease in the mean plasma clearance of theophylline. Patients who are receiving high dose theophylline or who are otherwise at increased risk of theophylline toxicity should be observed for signs of theophylline toxicity while receiving fluconazole, and therapy modified appropriately if signs of toxicity develop.
Tofacitinib.
Exposure of tofacitinib is increased when tofacitinib is co-administered with medications that result in both moderate inhibition of CYP3A4 and potent inhibition of CYP2C19 (e.g. fluconazole). Dosage adjustment of tofacitinib may be necessary.
Tolvaptan.
Exposure to tolvaptan is significantly increased (200% in AUC; 80% in Cmax) when tolvaptan, a CYP3A4 substrate, is co-administered with fluconazole, a moderate CYP3A4 inhibitor, with risk of significant increase in adverse effects particularly significant diuresis, dehydration and acute renal failure. In case of concomitant use, the tolvaptan dose should be reduced and the patient managed cautiously.
Vinca alkaloids.
Although not studied, fluconazole may increase the plasma levels of the vinca alkaloids (e.g. vincristine and vinblastine) and lead to neurotoxicity, which is possibly due to an inhibitory effect on CYP3A4.
Vitamin A.
Based on a case-report in one patient receiving combination therapy with all-transretinoid acid (an acid form of vitamin A) and fluconazole, central nervous system (CNS) related undesirable effects have developed in the form of pseudotumour cerebri, which disappeared after discontinuation of fluconazole treatment. This combination may be used but the incidence of CNS related undesirable effects should be borne in mind.
Voriconazole (CYP2C9, CYP2C19 and CYP3A4 inhibitor).
Concomitant administration of voriconazole and fluconazole at any dose is not recommended.
Warfarin.
A single dose of warfarin (15 mg) given to normal volunteers, following 14 days of orally administered fluconazole (200 mg) resulted in a 12% increase in the prothrombin time response (area under the prothrombin time-time curve). One of 13 subjects experienced a 2-fold increase in his prothrombin time response. In post-marketing experience, as with other azole antifungals, bleeding events (bruising, epistaxis, gastrointestinal bleeding, hematuria and melena) have been reported, in association with increases in prothrombin time in patients receiving fluconazole concurrently with warfarin. Careful monitoring of prothrombin time in patients receiving fluconazole and coumarin-type anticoagulants is recommended.
Zidovudine.
Fluconazole increases the Cmax and AUC of zidovudine, respectively, due to decrease in oral zidovudine clearance. The half-life of zidovudine was likewise prolonged following combination therapy with fluconazole. Patients receiving this combination should be monitored for the development of zidovudine-related adverse reactions. Dosage reduction of zidovudine may be considered.
Minor or no significant pharmacokinetic interactions that require no dosage adjustment. Oral contraceptives.
Oral contraceptives were administered as a single dose both before and after oral administration of fluconazole 50 mg once daily for 10 days in 10 healthy women. There was no significant difference in ethinyl estradiol or levonorgestrel AUC after the administration of 50 mg of fluconazole. The mean increase in ethinyl estradiol AUC was 6% (range: -47 to 108%) and levonorgestrel AUC increased 17% (range: -33 to 141%).
In a second study, twenty-five normal females received daily doses of either 200 mg fluconazole tablets or placebo for two, ten-day periods. The treatment cycles were one month apart with all subjects receiving fluconazole during one cycle and placebo during the other. Single doses of an oral contraceptive tablet containing levonorgestrel and ethinyl estradiol were administered on the final treatment day (day 10) of both cycles. Following administration of 200 mg of fluconazole, the mean percentage increase of AUC for levonorgestrel compared to placebo was 25% (range: -12 to 82%) and the mean percentage increase for ethinyl estradiol compared to placebo was 38% (range: -11 to 101%). Both of these increases were statistically significantly different from placebo.
In a third study, 21 healthy women received 300 mg weekly doses of fluconazole and single doses of ethinyl estradiol 35 microgram and norethindrone 0.5 mg. AUC of ethinyl estradiol was increased by 24% (range: 3 to 59%) and AUC of norethindrone was increased by 13% (range: -5 to 36%).
Multiple doses of fluconazole may increase exposure to hormone levels in women taking oral contraceptives and are unlikely to result in decreased efficacy of the oral contraceptive.
Two-way interactions.
Minor or no significant pharmacokinetic interactions that require no dosage adjustment. Azithromycin.
An open-label, randomized, three-way crossover study in 18 healthy subjects assessed the effect of a single 1200 mg oral dose of azithromycin on the pharmacokinetics of a single 800 mg oral dose of fluconazole as well as the effect of fluconazole on the pharmacokinetics of azithromycin. The estimated ratio of the mean AUC of fluconazole co-administered with azithromycin to fluconazole administered alone was 101%. The estimated ratio of the mean AUC of azithromycin co-administered with fluconazole to azithromycin alone was 107 %. The estimated ratio of the mean Cmax of fluconazole co-administered with azithromycin to fluconazole administered alone was 104%. The estimated ratio of the mean Cmax of azithromycin co-administered with fluconazole to azithromycin administered alone was 82%. See Table 2.

4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Fluconazole did not affect the fertility of male or female rats treated orally with daily doses of 5, 10 or 20 mg/kg or with parenteral doses of 5, 25 or 75 mg/kg, although the onset of parturition was slightly delayed at 20 mg/kg p.o. In an intravenous perinatal study in rats at 5, 20 and 40 mg/kg, dystocia and prolongation of parturition were observed in a few dams at 20 mg/kg and 40 mg/kg, but not at 5 mg/kg. The disturbances in parturition were reflected by a slight increase in the number of stillborn pups and decrease of neonatal survival at these dose levels. The effects on parturition in rats are consistent with the species-specific oestrogen-lowering property produced by high doses of fluconazole. Such a hormone change has not been observed in women treated with fluconazole (see Section 5.1 Pharmacodynamic Properties).
(Category D)
Drugs which have caused, are suspected to have caused or may be expected to cause an increased incidence of human foetal malformations or irreversible damage. These drugs may also have pharmacological effects. Accompanying texts should be consulted for further details.
There have been reports of spontaneous abortion and congenital abnormalities in infants whose mothers were treated with 150 mg of fluconazole as a single or repeated dose in the first trimester.
In one large observational cohort study, first trimester exposure to oral fluconazole was associated with a small increased risk of musculoskeletal malformations, corresponding to approximately 1 additional case per 1000 women treated with cumulative doses = 450 mg compared with women treated with topical azoles and to approximately 4 additional cases per 1000 women treated with cumulative doses over 450 mg. The adjusted relative risk was 1.29 (95% CI 1.05 to 1.58) for 150 mg oral fluconazole and 1.98 (95% CI 1.23 to 3.17) for doses over 450 mg fluconazole.
There are no adequate and well controlled studies in pregnant women. There have been reports of multiple congenital abnormalities in infants whose mothers were being treated for 3 or more months with high dose (400-800 mg/day) fluconazole therapy for coccidiomycosis. The relationship between fluconazole use and these events is unclear. Adverse foetal effects have been seen in animals only at high dose levels associated with maternal toxicity. These findings are not considered relevant to fluconazole used at therapeutic doses.
Case reports describe a distinctive and a rare pattern of birth defects among infants whose mothers received high dose (400 mg/kg to 800 mg/day) fluconazole during most or all of the first trimester of pregnancy. The features seen in these infants include: brachycephaly, abnormal facies, abnormal facies, abnormal calvarial development, cleft palate, femoral bowing, thin ribs and long bones, arthrogryposis and congenital heart disease.
Use in pregnancy should be avoided except in patients with severe or potentially life-threatening fungal infections in whom fluconazole may be used if the anticipated benefit outweighs the possible risk to the foetus. Effective contraceptive measures should be considered in women of child-bearing potential and should continue throughout the treatment period and for approximately 1 week (5 to 6 half-lives) after the final dose.
Fluconazole has been found in human breast milk at concentrations similar to plasma, hence its use in nursing mothers is not recommended. The elimination half-life from breast milk approximates the plasma elimination half-life of 30 hours. The estimated daily infant dose of fluconazole from breast milk (assuming mean milk consumption of 150 mL/kg/day) based on the mean peak milk concentration is 0.39 mg/kg/day, which is approximately 40% of the recommended neonatal dose (< 2 weeks of age) or 13% of the recommended infant dose for mucosal candidiasis. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for fluconazole and any potential adverse effects on the breastfed child from fluconazole or from the underlying maternal condition.
A pharmacokinetic study in 10 lactating women, who had temporarily or permanently stopped breast-feeding their infants, evaluated fluconazole concentrations in plasma and breast milk for 48 hours following a single 150 mg dose of fluconazole. Fluconazole was detected in breast milk at an average concentration of approximately 98% of those in maternal plasma. The mean peak breast milk concentration was 2.61 mg/L at 5.2 hours post-dose.4.7 Effects on Ability to Drive and Use Machines
When driving vehicles or operating machinery it should be taken into account that occasionally dizziness or seizures may occur.
4.8 Adverse Effects (Undesirable Effects)
Adults.
Summary of safety profile. Drug reaction with eosinophilia and systemic symptoms (DRESS) has been reported in association with fluconazole treatment (see Section 4.4 Special Warnings and Precautions for Use).
The safety profile of fluconazole appears similar in adults and children. The profile established for adults, given different dosage regimens and for different indications, is given below.
Multiple daily dosing for treatment of oral and for oral and oropharyngeal candidiasis; cryptococcal meningitis; or systemic candidiasis. Fluconazole is generally well tolerated. Sixteen percent of over 4000 patients treated in clinical trials of seven days or more experienced adverse events. Treatment was discontinued in 1.5% of patients due to adverse clinical events and in 1.3% due to laboratory abnormalities. Clinical adverse events were reported more frequently in HIV infected patients (21%) than in non-HIV infected patients (13%). However, the patterns in HIV infected and non-HIV infected patients were similar. The proportions of patients discontinuing therapy due to clinical adverse events were similar in the two groups (1.5%).
In some patients, particularly those with serious underlying diseases such as AIDS and cancer, changes in renal and haematological function test results and hepatic abnormalities have been observed during treatment with fluconazole and comparative agents, but the clinical significance and relationship to treatment is uncertain.
Hepatobiliary disorders.
In combined clinical trials and marketing experience, the spectrum of hepatic reactions has ranged from mild transient elevations in transaminases to clinical hepatitis, cholestasis and fulminant hepatic failure, including fatalities. Elevations in plasma levels of hepatic enzymes have been observed both in otherwise healthy patients and in patients with underlying disease (see Section 4.4 Special Warnings and Precautions for Use). There have been rare cases of serious hepatic reactions during treatment with fluconazole (see Section 4.4 Special Warnings and Precautions for Use). Instances of fatal hepatic reactions were noted to occur primarily in patients with serious underlying medical conditions (predominantly AIDS or malignancy) and often while taking multiple concomitant medications. In addition, transient hepatic reactions, including hepatitis and jaundice, have occurred among patients with no other identifiable risk factors. In each of these cases, liver function returned to baseline on discontinuation of fluconazole.
In two comparative trials evaluating the efficacy of fluconazole for the suppression of relapse of cryptococcal meningitis, a statistically significant increase was observed in median AST (SGOT) levels from a baseline value of 30 IU/L to 41 IU/L in one trial and 34 IU/L to 66 IU/L in the other. The overall rate of serum transaminase elevations of more than 8 times the upper limit of normal was approximately 1% in fluconazole-treated patients in the pre-marketing clinical trials which included patients with severe underlying disease, predominantly AIDS or malignancies, most of whom were receiving multiple concomitant medications, including many known to be hepatotoxic. The incidence of abnormally elevated serum transaminases was greater in patients taking fluconazole concomitantly with one or more of the following medications; rifampicin, phenytoin, isoniazid, valproic acid, or oral sulphonylurea hypoglycaemic agents.
Other adverse reactions observed include the following: see Table 3.
 See Table 4.
See Table 4.
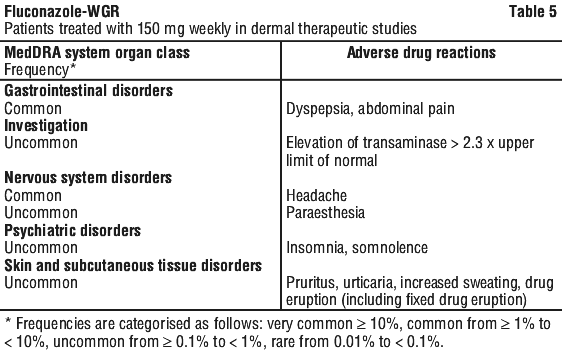
 See Table 5.
See Table 5.
Children.
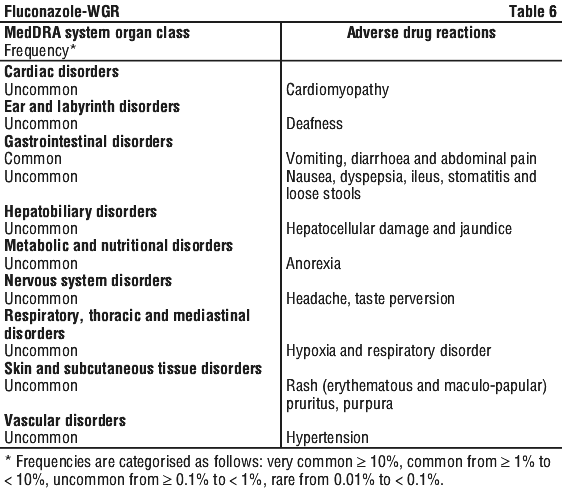
In clinical studies, 562 children, from birth to 17 years, received doses from 1 to 12 mg/kg per day, for up to 129 days. The majority of patients (n = 522) received 2 to 8 mg/kg per day for up to 97 days. Overall, approximately 10.3% experienced adverse events which were considered treatment related. The incidence of these adverse reactions and laboratory abnormalities do not suggest any marked difference between the paediatric population relative to the adult population. Based on this clinical trial data, the following adverse events were considered treatment related: see Table 6.
Post-marketing experience.
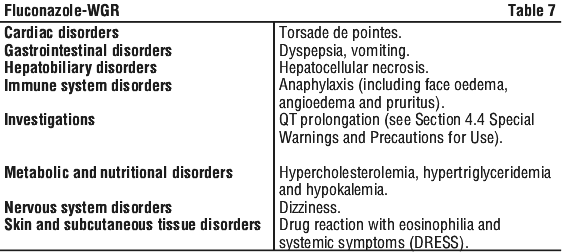
In addition the following adverse events have occurred during post-marketing: See Table 7.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal products is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.4.9 Overdose
The minimal lethal human dose has been not established. There have been reports of overdosage with fluconazole, and in one case, a 42-year-old patient infected with human immunodeficiency virus developed hallucinations and exhibited paranoid behaviour after reportedly ingesting 8,200 mg of fluconazole. The patient was admitted to hospital, and his condition resolved within 48 hours.
In the event of overdosage, symptomatic treatment (with supportive measures if necessary) should be undertaken.
Signs and symptoms are likely to be an extension of those under Section 4.8 Adverse Effects (Undesirable Effects).
There is no specific antidote. Treatment is symptomatic and supportive, including respiratory and cardiovascular function. Monitor for hypokalaemia and elevated liver enzymes; and obtain a full blood count to monitor for possible thrombocytopenia and agranulocytosis.
Fluconazole is largely excreted in the urine; forced volume diuresis would probably increase the elimination rate. A three-hour haemodialysis session decreases plasma levels by approximately 50%.
In mice and rats receiving very high doses of fluconazole, clinical effects, in both species, included decreased motility and respiration, ptosis, lacrimation, salivation, urinary incontinence, loss of righting reflex and cyanosis; death was sometimes preceded by clonic convulsions.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Fluconazole is a member of the bis-triazole class of antifungal agents. Fluconazole is a highly selective inhibitor of fungal cytochrome P-450 sterol C-14 alpha demethylation. Mammalian cell demethylation is much less sensitive to fluconazole inhibition. The subsequent loss of normal sterols correlates with the accumulation of 14 alpha-methyl sterols in fungi and may be responsible for the fungistatic activity of fluconazole. Fluconazole 50 mg daily given up to 28 days has been shown not to affect corticosteroid levels or ACTH stimulated response in healthy female volunteers. Plasma oestradiol levels and urinary free cortisol levels were decreased with little effect on plasma testosterone levels. Interaction studies with antipyrine indicate that single or multiple doses of fluconazole 50 mg do not affect its metabolism.
Susceptibility in vitro.
In vitro, fluconazole displays antifungal activity against clinically common Candida species (including C. albicans, C. parapsilosis, C. tropicalis). C. glabrata shows reduced susceptibility to fluconazole while C. krusei and C. auris are resistant to fluconazole. The minimum inhibitory concentrations (MICs) and epidemiological cut-off value (ECOFF) of fluconazole for C. guilliermondii are higher than for C. albicans.
Fluconazole also exhibits activity in vitro against Cryptococcus neoformans and Cryptococcus gattii as well as the endemic moulds Blastomyces dermatitidis, Coccidioides immitis, Histoplasma capsulatum and Paracoccidioides brasiliensis.
Pharmacokinetic/pharmacodynamic relationship.
In animal studies, there is a correlation between MIC values and efficacy against experimental mycoses due to Candida spp. In clinical studies, there is an almost 1:1 linear relationship between the AUC and the dose of fluconazole. There is also a direct though imperfect relationship between the AUC or dose and a successful clinical response of oral candidosis and to a lesser extent candidaemia to treatment. Similarly cure is less likely for infections caused by strains with a higher fluconazole MIC.
Mechanisms of resistance.
Candida spp have developed a number of resistance mechanisms to azole antifungal agents. Fungal strains which have developed one or more of these resistance mechanisms are known to exhibit high MICs to fluconazole which impacts adversely efficacy in vivo and clinically.
In usually susceptible species of Candida, the most commonly encountered mechanism of resistance development involves the target enzymes of the azoles, which are responsible for the biosynthesis of ergosterol. Resistance may be caused by mutation, increased production of an enzyme, drug efflux mechanisms, or the development of compensatory pathways.
There have been reports of superinfection with Candida species other than C. albicans, which often have inherently reduced susceptibility (C. glabrata) or resistance to fluconazole (e.g. C. krusei, C. auris). Such infections may require alternative antifungal therapy. The resistance mechanisms have not been completely elucidated in some intrinsically resistant (C. krusei) or emerging (C. auris) species of Candida.
Susceptibility testing breakpoints.
Based on analyses of pharmacokinetic/pharmacodynamic (PK/PD) data, susceptibility in vitro and clinical response EUCAST-AFST (European Committee on Antimicrobial susceptibility Testing-subcommittee on Antifungal Susceptibility Testing) has determined breakpoints for fluconazole for Candida species (EUCAST Fluconazole rationale document (2020)-version 3; European Committee on Antimicrobial Susceptibility Testing, Antifungal Agents, Breakpoint tables for interpretation of MICs, Version 10.0, valid from 2020-02-04). These have been divided into non-species related breakpoints; which have been determined mainly on the basis of PK/PD data and are independent of MIC distributions of specific species, and species related breakpoints for those species most frequently associated with human infection. These breakpoints are given in Table 8.
 For the most recent susceptibility testing interpretation according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST), Antifungal agents, please see (https://www.eucast.org/).
For the most recent susceptibility testing interpretation according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST), Antifungal agents, please see (https://www.eucast.org/).
Clinical trials.
No data available.
5.2 Pharmacokinetic Properties
Adults.
Absorption.
The pharmacokinetic properties of fluconazole are similar following administration by the intravenous or oral routes. In normal volunteers, the bioavailability of orally administered fluconazole is over 90% compared with intravenous administration. In fasted normal volunteers, peak plasma concentrations (Cmax) of 3.67/mL occurred in 4.02 hours (time to maximum serum concentration) after an oral dose of 200 mg fluconazole (Fluconazole-WGR) capsule.
Distribution.
Plasma concentrations are proportional to dose and steady-state levels are reached within 5-10 days with oral doses of 50-400 mg once daily. Steady-state levels are approximately 2.5 times the levels achieved with single doses. Administration of loading dose (on day 1) of twice the usual daily dose enables plasma levels to approximate to 90% steady-state levels by day 2. The apparent volume of distribution approximates to total body water. Plasma protein binding is low (11-12%).
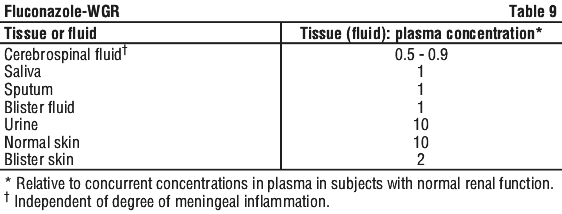
Fluconazole has been found to achieve good penetration into all tissues and body fluids studied. See Table 9.
Metabolism.
Following 100 mg dose of fluconazole a terminal plasma elimination half-life was reported to be approximately 30 hours (range 20 to 50 hours) in adults.
Excretion.
The major route of excretion is renal, with approximately 80% of the administered dose appearing in the urine as unchanged drug. About 11% of the dose is excreted in the urine as metabolites. The pharmacokinetics of fluconazole are markedly affected by reduction in renal function. There is an inverse relationship between the elimination half-life and creatinine clearance. The dose of fluconazole may need to be reduced in patients with impaired renal function (see Section 4.2 Dose and Method of Administration). A 3-hour haemodialysis session reduces plasma concentration by about 50%.
The long plasma elimination half-life provides the basis for single dose therapy for vaginal candidiasis, once daily and once weekly dosing for all other indications.
Children.
There are differences in the pharmacokinetics of fluconazole between adults and children, with children, after the neonatal period, generally having a faster elimination rate and larger volume of distribution than adults. These differences result in less accumulation on multiple dosing in children, with steady state achieved faster than in adults. Neonates have reduced elimination rates relative to adults and even higher volumes of distribution in comparison with older children. During the first 2 weeks after birth, the clearance of fluconazole increases (and the half-life is decreased) as renal function develops. The half-life obtained in infants was consistent with that found in older children, although the volume of distribution was higher. During the first year of life, the pharmacokinetics of fluconazole are similar to older children. No marked sex-related differences in pharmacokinetics are evident in children.
In children, the following mean pharmacokinetic data have been reported (see Table 10):
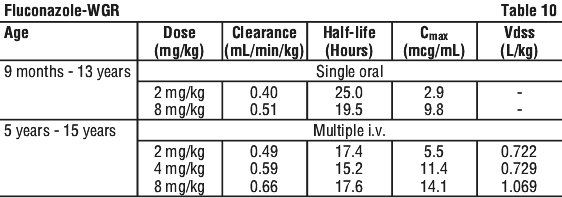 Clearance corrected for body weight was not affected by age in these studies. Mean body clearance in adults is reported to be 0.23 mL/min/kg.
Clearance corrected for body weight was not affected by age in these studies. Mean body clearance in adults is reported to be 0.23 mL/min/kg.
In premature newborns (gestational age 26 to 29 weeks), the mean clearance within 36 hours of birth was 0.180 mL/min/kg, which increased with time to a mean of 0.218 mL/min/kg 6 days later and 0.333 mL/min/kg 12 days later. Similarly, the half-life was 73.6 hours, which decreased with time to a mean of 53.2 hours 6 days later and 46.6 hours 12 days later.
Microbiology.
Fluconazole administered orally or intravenously was active in a variety of animal models of fungal infections using standard laboratory strains of fungi.
Fluconazole exhibits in vitro activity against Cryptococcus neoformans and Candida spp. Activity has been demonstrated in vivo in normal and immunocompromised animals against infections with Candida spp, including systemic candidiasis and in normal animals with C. neoformans, including intracranial infections. One case of cross-resistance of Candida to fluconazole in a patient (non-HIV) previously treated with ketoconazole has been reported.
The efficacy of fluconazole in vivo is greater than would be apparent from in vitro testing against the above-mentioned fungi.
Concurrent administration of fluconazole and amphotericin B in infected normal and immunocompromised mice showed antagonism of the two drugs in systemic infection with Aspergillus fumigatus. The clinical significance of results obtained in these studies is unknown.
5.3 Preclinical Safety Data
Genotoxicity.
Fluconazole, with or without metabolic activation, was negative in tests for mutagenicity in 4 strains of Salmonella typhimurium and in the mouse lymphoma system. Cytogenetic studies in vivo and in vitro showed no evidence of chromosomal mutations.
Carcinogenicity.
Fluconazole showed no evidence of carcinogenic potential in mice and rats treated orally for 24 months at doses of 2.5, 5 or 10 mg/kg/day (approximately 2-7 x recommended human dose). Male rats treated with 5 and 10 mg/kg/day had an increased incidence of hepatocellular adenomas.6 Pharmaceutical Particulars
6.1 List of Excipients
Fluconazole-WGR (fluconazole) capsules contain the following inactive ingredients: lactose monohydrate, maize starch, colloidal anhydrous silica, magnesium stearate, sodium lauryl sulfate and Tek Print SW-9008 Black Ink.
Capsule shell composition.
Patent blue, titanium dioxide, gelatin, erythrosine (100 mg and 200 mg only).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C.
6.5 Nature and Contents of Container
Fluconazole-WGR 50 mg, 100 mg and 200 mg.
The capsules are supplied in blister pack containing 28 capsules per pack.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.
 It is chemically designated as 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propanol.
It is chemically designated as 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propanol.
Its molecular formula is C13H12F2N6O and molecular weight is 306.3.
CAS number.
CAS number: 86386-73-4.7 Medicine Schedule (Poisons Standard)
S4.
Summary Table of Changes

