What is in this leaflet
This leaflet answers some common questions about Galafold®. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given Galafold® against the expected benefits it will have for you.
If you have any concerns about being given this medicine, ask your doctor.
Keep this leaflet. You may need to read it again.
What Galafold® is used for
Galafold® contains the active substance migalastat.
This medicine is used for the long- term treatment of Fabry disease in adults and adolescents aged 16 years and older who have certain genetic mutations (amenable mutations).
Fabry disease is caused by the lack of or a faulty enzyme called alpha- galactosidase A (α–Gal A).Depending upon the kind of mutation (change) in the gene that produces α– Gal A, the enzyme does not work properly or is completely absent. This enzyme defect leads to abnormal deposits of a fatty substance known as globotriaosylceramide (GL-3) leading to the symptoms of Fabry disease.
This medicine works by stabilising the enzyme that your body produces naturally, so that it can work better to reduce the amount of GL-3 that has accumulated in your cells and tissues.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
Before you take Galafold®
When you must not take Galafold®
- if you are allergic to migalastat or any of the other ingredients of this medicine (listed under What Galafold® contains)
You should not use this medicine after the expiry date printed on the Galafold® pack or if the packaging is torn or shows signs of tampering.
Before you start to take Galafold®
Talk to your doctor before taking Galafold® if you are currently taking enzyme replacement therapy.
You should not take Galafold® if you are also receiving enzyme replacement therapy.
Tell your doctor if you have any kidney problems.
If you have not told your doctor about any of the above, tell him/her before you take Galafold®.
Children and adolescents
This medicine has not been studied in children and adolescents under the age of 16 years; therefore, the effects in this age group are not known and use is not recommended.
Pregnancy, breast-feeding, andfertility
Pregnancy
There is very limited experience with the use of this medicine in pregnant women. If you are pregnant, think you may be pregnant, or are planning to have a baby, do not take this medicine until you have checked with your doctor, pharmacist, or nurse. It is not recommended to use Galafold® during pregnancy. While taking Galafold® you should use effective birth control.
Breast-feeding
Do not take this medicine if you are breast-feeding, until you have spoken with your doctor, pharmacist, or nurse. It is not yet known whether this medicine passes into human breast milk. Your doctor will decide whether you need to stop breast- feeding or temporarily stop your medicine.
Fertility
It is not yet known if this medicine affects fertility in both men or women.
If you are planning to have a baby, ask your doctor for advice.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket, health food shop, naturopath, herbalist or internet.
How to take Galafold®
Always take this medicine exactly as your doctor, pharmacist, or nurse has told you. Check with your doctor, pharmacist, or nurse if you are not sure.
Take one capsule every second day at the same time of the day.
Take the GALAFOLD® capsule on an empty stomach. Do not eat food at least 2 hours before and 2 hours after taking your medicine. Food may interfere with the absorption of the medicine. A minimum of 4 hours fasting around taking your medicine is needed to allow your medicine to be fully absorbed. Clear liquids can be consumed during this period.
How to take Galafold®
Swallow the capsule whole. Do not cut, crush, or chew the capsule.
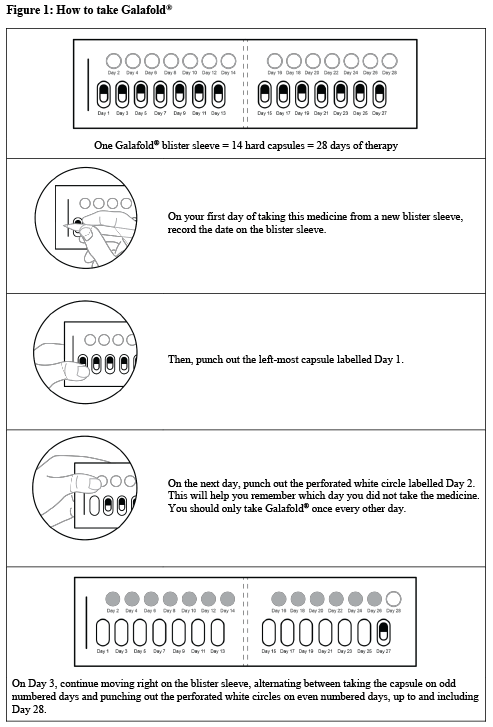
Refer to Figure 1, below, for further instructions on using the blister pack.
If you forget to take Galafold®
If you forget to take your capsule at the usual time but remember later that day, take the capsule when you remember on the same day.
If you miss a dose of this medicine for an entire day, do not take the missed capsule. Do not take Galafold capsules daily on consecutive days to make up for your missed dose. You should only take Galafold once every other day. Wait another 24 hours and take the next capsule at the time when you would usually take this medicine. Do not take two capsules together to make up for your missed dose.
If you stop taking Galafold®
Do not stop taking this medicine without talking to your doctor.
If you take more Galafold® than you should
If you take more capsules than you should, then you should stop taking the medicine and contact your doctor or pharmacist, or telephone the Poisons Information Centre on 13 11 26. You may get a headache and feel dizzy.
If you have any further questions on the use of this medicine, ask your doctor, pharmacist, or nurse.
While you are taking Galafold®
Things you must do
Keep all of your doctor’s appointments so your progress can be checked.
Your doctor may want to do some blood and other tests from time to time to check on your progress and to check for any unwanted side effects.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Galafold®.
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Do not be alarmed by the following list of side effects, you may not experience any of them.
Tell your doctor if you notice any of the following common side effects and they worry you:
Very common: may affect more than 1 in 10 people
- Headache
Common: may affect up to 1 in 10 people
- Palpitations (the feeling of a pounding heart)
- Sensation of spinning (vertigo)
- Diarrhoea
- Feeling sick (nausea)
- Stomach ache
- Constipation
- Dry mouth
- Sudden need to pass stools
- Indigestion (dyspepsia)
- Tiredness
- Raised levels of creatine phosphokinase in blood tests
- Weight gain
- Muscle spasms
- Muscle pain (myalgia)
- Painful stiff neck (torticollis)
- Tingling in extremities (paraesthesia)
- Dizziness
- Reduced sense of touch or sensation (hypoaesthesia)
- Depression
- Protein in the urine (proteinuria)
- Shortness of breath (dyspnoea)
- Nose bleed (epistaxis)
- Rash
- Persistent itch (pruritus)
Other side effects not listed above may occur in some people. Tell your doctor or pharmacist if you notice anything else that is making you feel unwell.
How to store Galafold®
Keep your capsules in the pack until it is time to take them. If you take the capsules out of the pack they may not keep well.
Keep your capsules in a cool dry place where the temperature stays below 30°C.
Do not store Galafold® or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and- a-half meteres above the ground is a good place to store medicines.
Disposal
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
Product description
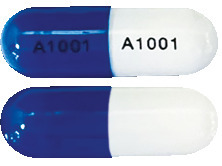
What Galafold® looks like and contents of the pack
Opaque, blue, and white hard capsules, marked with “A1001” in black ink, containing white to pale brown powder.
Galafold® is available in a blister pack containing 14 capsules.
What Galafold® contains
The active ingredient is migalastat. Each capsule contains migalastat hydrochloride equivalent to 123 mg migalastat
The inactive ingredients are:
Capsule contents: Pregelatinised maize starch and magnesium stearate
Capsule shell: Gelatin, titanium dioxide, and indigo carmine
Printing ink: Shellac, iron oxide black, and potassium hydroxide
Sponsor
Amicus Therapeutics Pty Ltd
21 Dorset Road
Northbridge, NSW 2063
Australia
Galafold® is a registered trademark of Amicus Therapeutics, Inc.
This leaflet was prepared in July 2018.
Copyright 2016. All rights reserved.
Published by MIMS December 2018














 In the ERT-naive trial, Galafold resulted in a statistically significant decrease in LVMi for all patients with amenable mutations (p < 0.05); the mean change from baseline in LVMi from Month 18 to 24 was -7.7 (95% CI: -15.4, -0.01; n = 27). After follow up in the open-label extension, the mean change from baseline in LVMi from Month 30 to 36 was -17.0 (95% CI: -26.2, -7.9; n = 15) (p < 0.05). The mean change from baseline in LVMi from Month 18 to 24 in patients with left ventricular hypertrophy at baseline (females with baseline LVMi > 95 g/m2 or males with baseline LVMi > 115 g/m2) was -18.6 (95% CI: -38.2, 1.0; n = 8). After follow up in the open-label extension, the mean change from baseline in LVMi in patients with left ventricular hypertrophy at baseline from Month 30 to 36 was -30.0 (95% CI: -57.9, -2.2; n = 4). No clinically significant differences in LVMi were observed during the initial 6-month placebo-controlled period.
In the ERT-naive trial, Galafold resulted in a statistically significant decrease in LVMi for all patients with amenable mutations (p < 0.05); the mean change from baseline in LVMi from Month 18 to 24 was -7.7 (95% CI: -15.4, -0.01; n = 27). After follow up in the open-label extension, the mean change from baseline in LVMi from Month 30 to 36 was -17.0 (95% CI: -26.2, -7.9; n = 15) (p < 0.05). The mean change from baseline in LVMi from Month 18 to 24 in patients with left ventricular hypertrophy at baseline (females with baseline LVMi > 95 g/m2 or males with baseline LVMi > 115 g/m2) was -18.6 (95% CI: -38.2, 1.0; n = 8). After follow up in the open-label extension, the mean change from baseline in LVMi in patients with left ventricular hypertrophy at baseline from Month 30 to 36 was -30.0 (95% CI: -57.9, -2.2; n = 4). No clinically significant differences in LVMi were observed during the initial 6-month placebo-controlled period.
 Australian Approved Name (AAN): migalastat hydrochloride.
Australian Approved Name (AAN): migalastat hydrochloride.