Notes
Distributed by Arrotex Pharmaceuticals
1 Name of Medicine
Ganvado denosumab (rch).
2 Qualitative and Quantitative Composition
Each vial contains a deliverable dose of 120 mg denosumab in 1.7 mL of solution (70 mg/mL).
Denosumab is a fully human IgG2 monoclonal antibody with high affinity and specificity for RANK ligand (RANKL). Denosumab has an approximate molecular weight of 147 kDa and is produced in genetically engineered mammalian cells (Chinese Hamster Ovary, CHO cells).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Ganvado is supplied as a sterile, preservative-free, clear, colourless to slightly yellow solution for subcutaneous (SC) injection at pH 5.2.
The solution may contain trace amounts of translucent to white proteinaceous particles.
4.1 Therapeutic Indications
Prevention of skeletal related events in patients with multiple myeloma and in patients with bone metastases from solid tumours.
Treatment of giant cell tumour of bone in adults or skeletally mature adolescents that is recurrent, or unresectable, or resectable but associated with severe morbidity.
Treatment of hypercalcaemia of malignancy that is refractory to intravenous bisphosphonate.
4.2 Dose and Method of Administration
Dosage (dose and interval).
The recommended dose of Ganvado for the prevention of skeletal related events is 120 mg administered as a single subcutaneous injection once every 4 weeks into the thigh, abdomen or upper arm.
The recommended dose of Ganvado for the treatment of giant cell tumour of bone and hypercalcaemia of malignancy is 120 mg administered as a single subcutaneous injection once every 4 weeks into the thigh, abdomen or upper arm with a loading dose of 120 mg on days 8 and 15 of the initial 4-week treatment period.
Daily supplementation with at least 500 mg calcium and 400 IU vitamin D is required in all patients, unless hypercalcaemia is present (see Section 4.4 Special Warnings and Precautions for Use, Vitamin supplementation and hypocalcaemia).
Method of administration.
Subcutaneous injection.
Administration should be performed by an individual who has been adequately trained in injection techniques.
Before administration, the Ganvado solution should be inspected for particulate matter and discolouration. Do not use if the solution is cloudy or discoloured. Do not shake excessively. To avoid discomfort at the site of injection, allow the vial to reach room temperature (up to 25°C) before injecting, and inject slowly. A 27-gauge needle or larger needle (e.g. 25-gauge) is recommended for the administration of Ganvado.
Product is for single-use in one patient only.
Inject the entire contents of the vial. Do not re-enter the vial.
Dispose of any medicinal product remaining in the vial.
Dosage adjustment.
Special populations. Use in elderly.
No dose adjustment is necessary in elderly patients (see Section 4.4 Special Warnings and Precautions for Use, Use in the elderly).
Renal impairment.
No dose adjustment is necessary in patients with renal impairment (see Section 4.4 Special Warnings and Precautions for Use, Use in renal impairment).
Use in paediatrics.
For treatment of giant cell tumour of bone in skeletally mature adolescents, the posology is the same as in adults.
Ganvado is not recommended in paediatric patients (age < 18) other than skeletally mature paediatric patients with giant cell tumour of bone (see Section 4.4 Special Warnings and Precautions for Use, Paediatric use).4.3 Contraindications
Pregnancy (see Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy).
Hypersensitivity to the active substance, to CHO-derived proteins or to any of the excipients (see Section 6.1 List of Excipients).
Severe untreated hypocalcaemia.
Unhealed lesions from dental or oral surgery.
4.4 Special Warnings and Precautions for Use
Vitamin supplementation and hypocalcaemia.
Pre-existing hypocalcaemia must be corrected prior to initiating therapy with Ganvado.
Supplementation with calcium and vitamin D is required in all patients unless hypercalcaemia is present.
Hypocalcaemia can occur at any time during therapy with Ganvado. Monitoring of calcium levels should be conducted (i) prior to the initial dose of Ganvado, (ii) within two weeks after the initial dose, (iii) if suspected symptoms of hypocalcaemia occur (see Section 4.8 Adverse Effects (Undesirable Effects) for symptoms). Additional monitoring of calcium level should be considered during therapy in patients with risk factors for hypocalcaemia, or if otherwise indicated based on the clinical condition of the patient. Patients should be encouraged to report symptoms indicative of hypocalcaemia.
In the post-marketing setting, severe symptomatic hypocalcaemia (including fatal cases) has been reported (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience), with most cases occurring in the first weeks of initiating therapy, but can occur later.
If hypocalcaemia occurs while receiving Ganvado, additional short term calcium supplementation may be necessary (see Section 4.4 Special Warnings and Precautions for Use, Use in renal impairment; Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
Use in hepatic impairment.
The safety and efficacy of Ganvado has not been studied in patients with hepatic impairment.
Use in renal impairment.
No dose adjustment is necessary in patients with renal impairment.
In clinical studies of patients without advanced cancer, but with varying degrees of renal function (including patients with severe renal impairment [creatinine clearance < 30 mL/min] or receiving dialysis) there was a greater risk of developing hypocalcaemia with increasing degree of renal impairment, and in the absence of calcium supplementation. Monitoring calcium levels and adequate intake of calcium and vitamin D is important in patients with severe renal impairment or receiving dialysis (see Section 4.4 Special Warnings and Precautions for Use, Vitamin supplementation and hypocalcaemia).
Osteonecrosis of the jaw.
Osteonecrosis of the jaw (ONJ) has occurred in patients treated with denosumab, with the majority of cases occurring within 5 months after the last dose. In clinical trials, the incidence of ONJ was higher with longer duration of exposure (see Section 4.8 Adverse Effects (Undesirable Effects)).
Patients who developed ONJ in clinical studies generally had known risk factors for ONJ, including invasive dental procedures (e.g. tooth extraction, dental implants, oral surgery), poor oral hygiene or other pre-existing dental disease, local gum or oral infection, advanced malignancies, or concomitant therapies (e.g. chemotherapy, corticosteroids, angiogenesis inhibitors). An oral examination should be performed by the prescriber prior to initiation of Ganvado treatment and a dental examination with appropriate preventive dentistry is recommended prior to treatment with Ganvado, especially in patients with risk factors for ONJ. Good oral hygiene practices should be maintained during treatment with Ganvado.
Patients should avoid invasive dental procedures during treatment with Ganvado. For patients in whom invasive dental procedures cannot be avoided, the clinical judgement of the treating physician should guide the management plan of each patient based on individual benefit-risk assessment. Patients who are suspected of having or who develop ONJ while on Ganvado should receive care by a dentist or an oral surgeon. If ONJ occurs during treatment with Ganvado a temporary interruption of treatment should be considered based on individual benefit-risk assessment until the condition resolves.
Atypical femoral fractures.
Atypical femoral fracture has been reported with Ganvado (see Section 4.8 Adverse Effects (Undesirable Effects)). Atypical femoral fractures may occur with little or no trauma in the subtrochanteric and diaphyseal regions of the femur and may be bilateral. Specific radiographic findings characterise these events. Atypical femoral fractures have also been reported in patients with certain co-morbid conditions (e.g. vitamin D deficiency, rheumatoid arthritis, hypophosphatasia) and with use of certain pharmaceutical agents (e.g. bisphosphonates, glucocorticoids, proton pump inhibitors).
These events have also occurred without antiresorptive therapy. During Ganvado treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Patients presenting with such symptoms should be evaluated for an incomplete femoral fracture, and the contralateral femur should also be examined.
Hypercalcaemia following treatment discontinuation in patients with giant cell tumour of bone and in patients with growing skeletons.
Clinically significant hypercalcaemia requiring hospitalisation and complicated by acute renal injury has been reported in Ganvado-treated patients with giant cell tumour of bone and patients with growing skeletons weeks to months following treatment discontinuation. After treatment is discontinued, monitor patients for signs and symptoms of hypercalcaemia, consider periodic assessment of serum calcium as clinically indicated, and treat appropriately. Re-evaluate the patient's calcium and vitamin D supplementation requirements. Manage hypercalcaemia as clinically appropriate (see Section 4.4 Special Warnings and Precautions for Use, Paediatric use; Section 4.8 Adverse Effects (Undesirable Effects)).
Multiple vertebral fractures (MVF) following treatment discontinuation.
Multiple vertebral fractures (MVF), not due to bone metastases, may occur following discontinuation of treatment with Ganvado, particularly in patients with risk factors such as osteoporosis or prior fractures.
Advise patients not to interrupt Ganvado therapy without their physician's advice. When Ganvado treatment is discontinued, evaluate the individual patient's risk for vertebral fractures (see Section 4.8 Adverse Effects (Undesirable Effects)).
Drugs with same active ingredient.
Ganvado contains the same active ingredient found in Prolia (denosumab), used for the treatment of postmenopausal osteoporosis. Patients being treated with Ganvado should not be treated with Prolia and/or other denosumab-containing medicines concomitantly.
Warnings for excipients.
Patients with rare hereditary problems of fructose intolerance should not use Ganvado.
Use in the elderly.
Of the total number of patients in clinical studies in patients with advanced cancer, 1260 patients (44.4%) treated with Ganvado were ≥ 65 years old. No overall differences in safety or efficacy were observed between these patients and younger patients.
Paediatric use.
The safety and efficacy of Ganvado in paediatric patients (age < 18) have not been established other than skeletally mature adolescents with giant cell tumour of bone. Ganvado is not recommended for use in paediatric patients other than skeletally mature adolescents with giant cell tumour of bone. Clinically significant hypercalcaemia after treatment discontinuation has been reported in the post-marketing setting in paediatric patients with growing skeletons who received denosumab for giant cell tumour of bone or for unapproved indications (see Section 4.4 Special Warnings and Precautions for Use, Hypercalcaemia following treatment discontinuation in patients with giant cell tumour of bone and in patients with growing skeletons).
In Study 20062004, Ganvado has been evaluated in a subset of 28 adolescent patients (aged 13-17 years) with giant cell tumour of bone who had reached skeletal maturity defined by at least 1 mature long bone (e.g. closed epiphyseal growth plate of the humerus) and body weight ≥ 45 kg. Efficacy results in skeletally mature adolescents appeared to be similar to those observed in adults (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Adolescent primates had abnormal growth plates when administered denosumab at doses of 10 mg/kg and higher, which resulted in exposures up to 2.8 times those observed in adult humans dosed at 120 mg subcutaneously every 4 weeks based on area under the curve (AUC). In neonatal cynomolgus monkeys exposed in utero to denosumab at 50 mg/kg, there was increased postnatal mortality; abnormal bone growth resulting in reduced bone strength, reduced haematopoiesis, and tooth malalignment; absence of peripheral lymph nodes; decreased neonatal growth and other adverse effects (see Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy). In neonatal rats, inhibition of RANKL (target of denosumab therapy) with a construct of osteoprotegerin bound to Fc (OPG-Fc) was associated with inhibition of bone growth and tooth eruption and lower body weight gain. These changes were partially reversible when dosing of RANKL inhibitor was discontinued. Therefore, treatment with denosumab may impair bone growth in children with open growth plates and may inhibit eruption of dentition.
Effects on laboratory tests.
No interactions with laboratory and diagnostic tests have been identified.4.5 Interactions with Other Medicines and Other Forms of Interactions
No drug-drug interaction studies have been conducted.
In clinical studies, Ganvado has been administered in combination with standard anticancer treatment and in patients previously receiving bisphosphonates.
The pharmacokinetics and pharmacodynamics of denosumab were not altered by concomitant chemotherapy and/or hormone therapy nor by previous IV bisphosphonate exposure.
Denosumab should not be administered concomitantly with bisphosphonates.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No data are available on the effect of denosumab on human fertility. Denosumab had no effect on female fertility or male reproductive organs or sperm motility in cynomolgus monkeys at subcutaneous doses up to 12.5 mg/kg/week (females) or 50 mg/kg/month (males), yielding exposures that were approximately 15-fold higher than the human exposure at 120 mg subcutaneous administered once every month.
(Category D)
There are no adequate and well-controlled studies of Ganvado in pregnant women. Ganvado is contraindicated for use during pregnancy. Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of Ganvado. Any effects of Ganvado are likely to be greater during the second and third trimesters of pregnancy since monoclonal antibodies are transported across the placenta in a linear fashion as pregnancy progresses, with the largest amount transferred during the third trimester. Inform the patient of the potential hazard to a foetus if the patient becomes pregnant while exposed to Ganvado.
Developmental toxicity studies have been performed with denosumab in cynomolgus monkeys and have shown serious adverse effects on development (including foetal and infant lethality). Denosumab was shown to cross the placenta in monkeys.
In a study of cynomolgus monkeys with denosumab at 12.5 mg/kg/week given during the period equivalent to the first trimester at AUC exposures up to 10-fold higher than the human dose (120 mg every 4 weeks), there was no evidence of maternal or foetal harm. In this study, foetal lymph nodes were not examined.
In another study of cynomolgus monkeys with denosumab throughout pregnancy at 50 mg/kg/month, yielding AUC exposures 12-fold higher than the human dose (120 mg every 4 weeks), there were increased stillbirths and postnatal mortality; abnormal bone growth resulting in reduced bone strength, almost complete obliteration of bone marrow spaces (leading to reduced haematopoiesis), and tooth malalignment, dental dysplasia and shortened/straighter dental arch (although no effect on the pattern or date of tooth eruption); altered appearance of eyes (increased apparent size, exophthalmos); absence of peripheral lymph nodes; and decreased neonatal growth. Following a 6 month period after birth, bone-related changes showed incomplete recovery. The effects on lymph nodes, tooth malalignment and dental dysplasia persisted, and minimal to moderate mineralisation in multiple tissues was seen in one animal. There was no evidence of maternal harm prior to labour; adverse maternal effects occurred infrequently during labour. Maternal mammary gland development was normal. A no observed adverse effect level has not been established in animal studies and the findings are attributable to the primary pharmacological activity of denosumab.
Preclinical studies in RANK/RANKL-knockout mice suggest absence of RANKL could interfere with the development of lymph nodes in the foetus. Knockout mice lacking RANK or RANKL also exhibited decreased body weight, reduced bone growth and a lack of tooth eruption. Similar phenotypic changes (inhibition of bone growth and tooth eruption) were observed in a study in neonatal rats using a surrogate for denosumab, the RANKL inhibitor osteoprotegerin bound to Fc (OPG-Fc). These changes were partially reversible when dosing of RANKL inhibitor was discontinued. Therefore, treatment with denosumab may impair bone growth in children with open growth plates and may inhibit eruption of dentition.
Preclinical studies in RANK/RANKL-knockout mice suggest absence of RANKL during pregnancy may interfere with maturation of the mammary gland leading to impaired lactation post-partum.
It is unknown whether denosumab is excreted in human milk. Only limited excretion of denosumab in milk was observed in a study in monkeys. A decision on whether to abstain from breast-feeding or to abstain from therapy with Ganvado should be made, taking into account the benefit of breast-feeding to the newborn/infant and the benefit of Ganvado therapy to the woman.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive or use machinery have been performed.
4.8 Adverse Effects (Undesirable Effects)
Bone metastasis from solid tumours.
Data from three active-controlled multicentre trials were used for the safety analysis in 5677 patients with bone metastases from either prostate cancer, breast cancer, other solid tumours or patients with multiple myeloma (all patients with advanced cancer). A total of 2841 patients were exposed to 120 mg of Ganvado administered once every 4 weeks as a single subcutaneous injection, and 2836 patients were exposed to 4 mg (dose-adjusted for reduced renal function) of zoledronic acid administered once every 4 weeks as an IV infusion. The median (Q1, Q3) duration of exposure to Ganvado for the safety analysis was 12 months (6, 18) for prostate cancer, 17 months (10, 21) for breast cancer, and 7 months (4, 14) for other solid tumours and multiple myeloma. See Table 1.
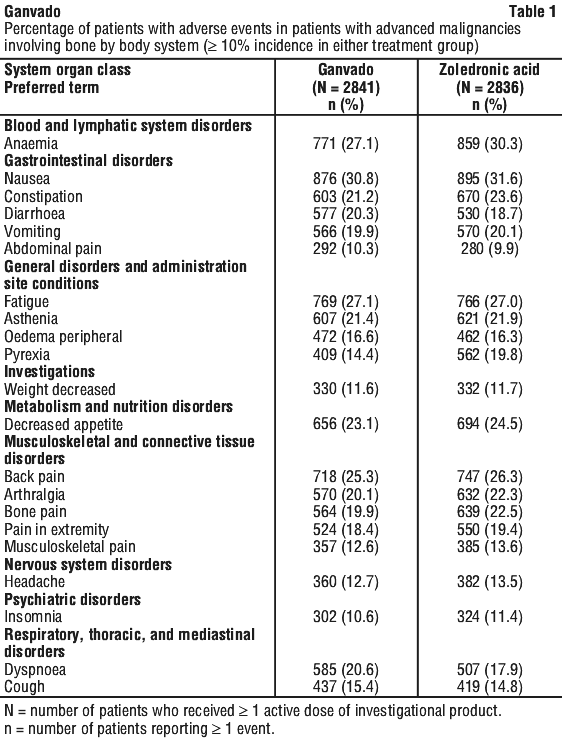 Hypophosphataemia has been reported as a common adverse drug reaction.
Hypophosphataemia has been reported as a common adverse drug reaction.
Giant cell tumour of bone.
The safety of Ganvado was evaluated in two phase 2 open-label, single arm studies in which a total of 548 patients with giant cell tumour of bone received at least 1 dose of Ganvado. Patients received 120 mg Ganvado subcutaneously every 4 weeks with a loading dose of 120 mg on days 8 and 15 of the initial 4-week period. Of the 548 patients who received Ganvado, 467 patients were treated with Ganvado for ≥ 1 year, 323 patients for ≥ 2 years, 255 patients for ≥ 3 years, 195 patients for ≥ 4 years and 149 patients for ≥ 5 years. The median (Q1, Q3) number of doses received was 33.0 (17.0, 63.0); the minimum number of doses received was 4 and the maximum was 138. The median (Q1, Q3) number of months on study was 59.61 (28.52, 79.61). The median (range) age was 33 (13 to 83) years; 28 patients were skeletally mature adolescents (aged 13 to 17 years).
The overall safety and tolerability profile of Ganvado in patients with giant cell tumour of bone was similar to that reported in trials of patients with bone metastases from solid tumours. For skeletally mature adolescent patients with GCTB, the safety profile appears to be similar to that in adult patients with GCTB.
The most common adverse reactions in patients with giant cell tumour of bone receiving Ganvado (per-patient incidence greater than or equal to 20%) were arthralgia, back pain, pain in extremity, fatigue, headache and, nausea.
Hypercalcaemia following treatment discontinuation in patients with giant cell tumour of bone has been observed uncommonly.
Hypercalcaemia of malignancy.
The safety of Ganvado was evaluated in an open-label, single-arm trial (Study 20070315) in which 33 patients were enrolled with hypercalcaemia of malignancy (with or without bone metastases) refractory to treatment with intravenous bisphosphonate. Patients received Ganvado subcutaneously every 4 weeks with additional 120 mg doses on Days 8 and 15 of the initial 4-week period. Entry criteria included patients who had refractory hypercalcaemia of malignancy (defined as an albumin-corrected calcium of > 12.5 mg/dL [3.1 mmol/L] despite treatment with intravenous bisphosphonate in the last 7-30 days).
Patients receiving dialysis for renal failure or who had treatment with thiazides, calcitonin, mithromycin, or gallium nitrate within their window of expected therapeutic effect prior to the date of screening corrected serum calcium (CSC) were excluded. During the trial, serum calcium was collected every few days in the first month, weekly during the second month, and monthly thereafter.
Of the 33 patients who received Ganvado, 33 patients were treated with Ganvado for ≥ 1 month, 5 patients for ≥ 6 months, and 3 patients for ≥ 1 year. The median number of doses received was 4 (range: 1 to 25 doses) and the median number of months on study was 1.8 (range: 0 to 23 months). Sixty-four percent of enrolled patients were men and 70% were white. The median age was 63 years (range: 22 to 89 years).
The adverse reaction profile of Ganvado in patients with hypercalcaemia of malignancy was similar to that reported in patients with bone metastases from solid tumours and giant cell tumour of bone.
The most common adverse reactions were nausea, dyspnoea, decreased appetite, headache, peripheral oedema, and vomiting. No adverse events leading to discontinuation were reported as related to Ganvado treatment.
Hypocalcaemia.
In three phase 3 active-controlled clinical trials in patients with advanced malignancies involving bone, hypocalcaemia was reported in 9.6% of patients treated with Ganvado and 5.0% of patients treated with zoledronic acid. A decrease in serum calcium levels to the range between 1.5 to 1.75 mmol/L was experienced in 2.5% of patients treated with Ganvado and 1.2% of patients treated with zoledronic acid. A decrease in serum calcium levels to < 1.5 mmol/L was experienced in 0.6% of patients treated with Ganvado and 0.2% of patients treated with zoledronic acid.
In a phase 3 active-controlled clinical trial in patients with newly diagnosed multiple myeloma, hypocalcaemia was reported in 16.9% of patients treated with Ganvado and 12.4% of patients treated with zoledronic acid. A decrease in serum calcium levels to the range between 1.5 to 1.75 mmol/L was experienced in 1.4% of patients treated with Ganvado and 0.6% of patients treated with zoledronic acid. A decrease in serum calcium levels to the range between 0.8 to 1.5 mmol/L was experienced in 0.4% of patients treated with Ganvado and 0.1% of patients treated with zoledronic acid.
In two phase 2 open-label trials in patients with giant cell tumour of bone, hypocalcaemia was reported in 5.7% of patients. None of the adverse events was considered serious.
In a phase 2 open-label, single-arm trial in patients with hypercalcaemia of malignancy refractory to intravenous bisphosphonate, hypocalcaemia was reported in 9.1% of patients treated with Ganvado.
Osteonecrosis of the jaw (ONJ).
In the primary treatment phase of three phase 3 active-controlled clinical trials in patients with advanced malignancies involving bone, ONJ was confirmed in 1.8% of patients treated with Ganvado (median exposure of 12 months; range 0.1 to 40.5) and 1.3% of patients treated with zoledronic acid. Clinical characteristics of these cases were similar between treatment groups.
Among patients with confirmed ONJ, most had a history of tooth extraction, poor oral hygiene, and/or use of a dental appliance. In addition, most patients were receiving or had received chemotherapy. The trials in patients with breast or prostate cancer included a pre-specified Ganvado extension treatment phase (median overall exposure of 14.9 months; range 0.1 - 67.2) where patients were offered open label Ganvado. The patient-year adjusted incidence of confirmed ONJ was 1.1 per 100 patient-years during the first year of treatment, 3.7 in the second year and 4.6 thereafter. The median time to ONJ was 20.6 months (range: 4 - 53).
In a phase 3 double-blind, active-controlled clinical trial in patients with newly diagnosed multiple myeloma, ONJ was confirmed in 4.1% of patients in the Ganvado group (median exposure of 15.8 months; range 1 - 49.8) and 2.8% of patients in the zoledronic acid group. At the completion of the double-blind treatment phase of this trial, the patient-year adjusted incidence of confirmed ONJ in the Ganvado group (median exposure of 19.4 months; range 1 - 52) was 2.0 per 100 patient-years during the first year of treatment, 5.0 in the second year, and 4.5 thereafter. The median time to ONJ was 18.7 months (range: 1 - 44).
In a phase 3 placebo-controlled clinical trial with an extension treatment phase evaluating Ganvado for the prevention of bone metastases in patients with non-metastatic prostate cancer (a patient population for which Ganvado is not indicated), with longer treatment exposure of up to 7 years, the patient-year adjusted incidence of confirmed ONJ was 1.1 per 100 patient-years during the first year of treatment, 3.0 in the second year, and 7.1 thereafter.
In two phase 2 open-label studies in patients with giant cell tumour of bone, ONJ occurred in 4 of 304 (1.3%) of patients. The median time to ONJ was 16 months (range 13-20).
In a phase 2 open-label, single-arm trial in patients with hypercalcaemia of malignancy refractory to intravenous bisphosphonate, no cases of ONJ were reported.
In a phase 2 open-label clinical trial in patients with giant cell tumour of bone (study 20062004), ONJ was confirmed in 6.8% of patients (median number of 34 doses; range 4 - 116). At the completion of the trial, median time on trial including safety follow-up phase was 60.9 months (range: 0 - 112.6). The patient-year adjusted incidence of confirmed ONJ was 1.5 per 100 patient-years overall (0.2 per 100 patient-years during the first year of treatment, 1.5 in the second year, 1.8 in the third year, 2.1 in the fourth year, 1.4 in the fifth year, and 2.2 thereafter). The median time to ONJ was 41 months (range: 11 - 96).
Study 20140114 was conducted to continue to follow patients with GCTB who were treated in study 20062004 for an additional 5 or more years. ONJ was reported in 6 patients (11.8%) of the 51 exposed patients with median total 42 doses of denosumab. Three of these cases of ONJ were medically confirmed.
Atypical femoral fractures (AFF).
Atypical femoral fracture has been reported uncommonly in patients treated with Ganvado and the risk increased with longer duration of treatment. Events have occurred during treatment and up to 9 months after treatment was discontinued.
In the clinical trial programme for GCTB, atypical femoral fractures have been reported commonly in patients treated with Ganvado. In study 20062004, incidence of confirmed AFF was 0.95% (5/526) in patients with GCTB. In the follow up study 20140114, the incidence of confirmed AFF was 3.9% (2/51) of patients exposed to denosumab.
Paediatric patients.
The safety profile of Ganvado in 28 skeletally mature adolescent patients with giant cell tumour of bone was consistent with that in adult patients.
Drug hypersensitivity events.
In clinical trials in patients with advanced cancer, drug hypersensitivity events were reported in 0.9% and 0.4% of patients treated with Ganvado and zoledronic acid, respectively.
Pancreatitis.
In a randomised controlled trial in postmenopausal women with osteoporosis receiving 60 mg denosumab or placebo once every 6 months, pancreatitis was reported in 8 patients (0.2%) in the denosumab and 4 patients (0.1%) in the placebo groups. An increased incidence has not been observed in randomised controlled trials in the oncology setting.
Hypercalcaemia.
Hypercalcaemia has been observed following treatment discontinuation in patients with growing skeletons (a patient population for which Ganvado is not indicated).
Multiple vertebral fractures.
Multiple vertebral fractures, not due to bone metastases, have occurred in patients with risk factors such as osteoporosis or prior fractures following treatment discontinuation.
Post-marketing experience.
The following adverse reactions have been identified during post approval use of Ganvado:
Rare events of severe symptomatic hypocalcaemia (including fatal cases) have been reported in patients at increased risk of hypocalcaemia. Examples of the clinical manifestations of severe symptomatic hypocalcaemia have included QT interval prolongation, tetany, seizures and altered mental status (see Section 4.4 Special Warnings and Precautions for Use, Vitamin supplementation and hypocalcaemia). Symptoms of hypocalcaemia in denosumab clinical studies included paraesthesias or muscle stiffness, twitching, spasms and muscle cramps.
Hypersensitivity, including anaphylactic reactions.
Musculoskeletal pain, including severe cases.
Uncommon events of injection site reactions (including injection site pain) have been observed.
Lichenoid drug eruptions (e.g. lichen planus-like reactions) have been observed uncommonly.
Alopecia has been observed commonly.
There have been reports of osteonecrosis of the external auditory canal in patients using denosumab.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.4.9 Overdose
There is no experience with overdosage with Ganvado. Ganvado has been administered in clinical studies using doses up to 180 mg every 4 weeks (cumulative doses up to 1080 mg over 6 months), and 120 mg weekly for 3 weeks.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Bone metastasis from solid tumours.
RANKL exists as a transmembrane or soluble protein. RANKL is essential for the formation, function and survival of osteoclasts, the sole cell type responsible for bone resorption. Increased osteoclast activity, stimulated by RANKL, is a key mediator of bone destruction in bone disease in metastatic tumours and multiple myeloma. Denosumab binds with high affinity and specificity to RANKL, preventing RANKL from activating its only receptor, RANK, on the surface of osteoclasts and their precursors. Prevention of RANKL-RANK interaction results in reduced osteoclast numbers and function, and thereby decreases bone resorption and cancer-induced bone destruction.
RANKL inhibition resulted in reduced bone lesions and delayed formation of de novo bone metastases in some nonclinical models. RANKL inhibition reduced skeletal tumour growth and this effect was additive when combined with other anticancer therapies.
Giant cell tumour of bone.
Giant cell tumours of bone are characterised by stromal cells expressing RANKL and osteoclast-like giant cells expressing RANK. In patients with giant cell tumour of bone, denosumab binds to RANKL, significantly reducing or eliminating osteoclast-like giant cells. Consequently, osteolysis is reduced and proliferative tumour stroma can be replaced with non-proliferative, differentiated, woven new bone which may show an increase in density.
Hypercalcaemia of malignancy refractory to intravenous bisphosphonates.
The primary aetiology of both skeletal and humoral hypercalcaemia of malignancy is increased bone resorption, which leads to elevated calcium concentrations in the extracellular fluid. The increase in bone resorption is initiated by the release of signalling molecules such as PTHrP, prostaglandins, and cytokine by malignant and stromal cells. These molecules stimulate osteoblasts and other stromal cells to express RANKL, which upon binding its receptor RANK upregulates osteoclast recruitment and differentiation and thus bone resorption, with a resultant increase in calcium concentrations of the extracellular fluid and serum. Ganvado binds to RANKL preventing RANK/RANKL mediated osteoclast formation, function, and survival thereby lowering serum calcium levels.
Pharmacodynamics.
In a phase 2 study of IV-bisphosphonate naïve patients with breast cancer and bone metastases, subcutaneous (SC) doses of Ganvado 120 mg every 4 weeks (Q4W), caused a rapid reduction in the markers of bone resorption: urinary N-telopeptide corrected for creatinine (uNTx/Cr) and serum C-telopeptide (sCTx) with median reduction of 82% for uNTx/Cr within 1 week. Reductions in bone resorption markers were maintained, with median uNTx/Cr reductions of 74% to 82% from weeks 2 to 25 of continued 120 mg Q4W dosing. Median reduction of approximately 80% in uNTx/Cr from baseline after 3 months of treatment were also observed across 2075 Ganvado-treated advanced cancer patients (breast, prostate, multiple myeloma or other solid tumours) naïve to IV-bisphosphonate in the phase 3 clinical trials.
Similarly, in a phase 2 study of patients with advanced malignancies and bone metastases (including patients with multiple myeloma and bone disease) who were receiving intravenous bisphosphonate therapy, yet had uNTx/Cr levels > 50 nanoM/mM, SC dosing of Ganvado administered either every 4 weeks or every 12 weeks caused an approximate 80% reduction in uNTx/Cr from baseline after 3 and 6 months of treatment. Overall, 97% of patients in the Ganvado groups had at least one uNTx/Cr value < 50 nanoM/mM up to week 25 of the study.
In a phase 3 study of patients with newly diagnosed multiple myeloma who received SC doses of Ganvado 120 mg every 4 weeks (Q4W), median reductions in uNTx/Cr of approximately 75% were observed by week 5. Reductions in bone turnover markers were maintained, with median reductions of 74% to 79% for uNTx/Cr from weeks 9 to 49 of continued 120 mg Q4W dosing.
In a phase 2 study of patients with giant cell tumour of bone who received subcutaneous doses of Ganvado 120 mg every 4 weeks (Q4W) with loading doses on days 8 and 15 of the initial 4-week treatment period, median reductions in uNTx/Cr and sCTx of approximately 80% were observed by week 9. Reductions in bone turnover markers were maintained, with median reductions of 56% to 77% for uNTx/Cr and 79% to 83% for sCTx from weeks 5 to 25 of continued 120 mg Q4W dosing.
Clinical trials.
Clinical efficacy in patients with bone metastases from solid tumours. Efficacy and safety of 120 mg Ganvado subcutaneously every 4 weeks or 4 mg zoledronic acid (dose-adjusted for reduced renal function) IV every 4 weeks were compared in three randomised, double blind, active controlled studies, in IV-bisphosphonates naïve patients with advanced malignancies involving bone. A total of 2,046 adults with breast cancer with at least one bone metastasis (Study 20050136), 1,776 adults with other solid tumours (including non-small cell lung cancer, renal cell cancer, colorectal cancer, small cell lung cancer, bladder cancer, head and neck cancer, gastrointestinal/genitourinary cancer and others, excluding breast and prostate cancer) with at least one bone metastasis or multiple myeloma (Study 20050244), and 1,901 men with castrate-resistant prostate cancer with at least one bone metastasis (Study 20050103) were included. The primary and secondary endpoints evaluated the occurrence of one or more skeletal related events (SREs) defined as any of the following: pathologic fracture, radiation therapy to bone, surgery to bone or spinal cord compression.
Ganvado reduced the risk of developing a SRE, or developing multiple SREs (first and subsequent) in patients with advanced malignancies involving bone (see Figure 1 and Table 2).
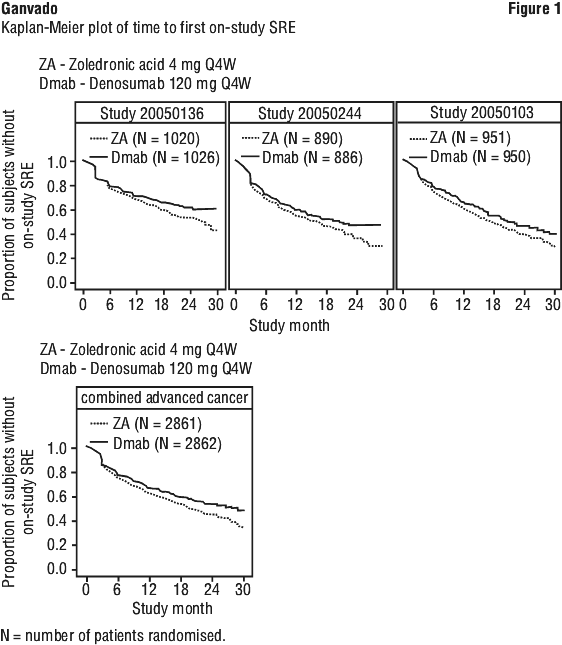
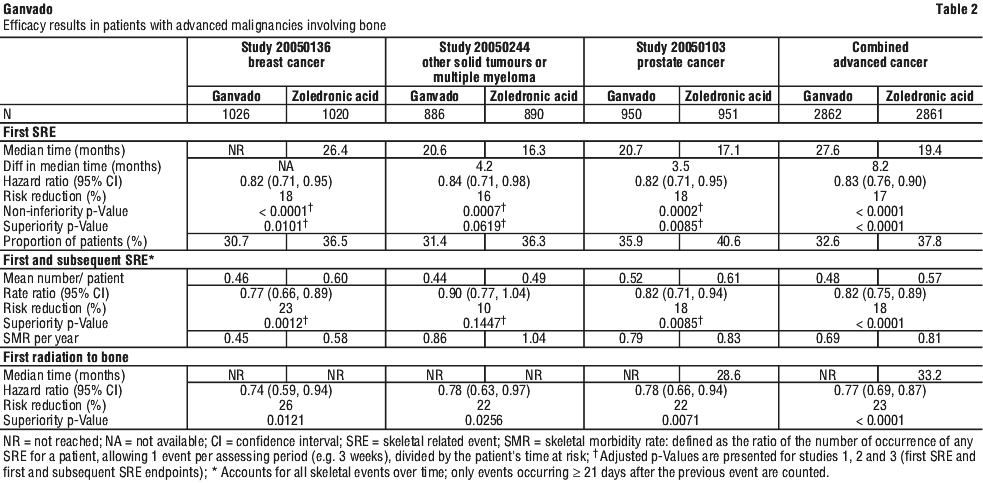 In a post hoc analysis of Study 20050244 (including solid tumours, excluding multiple myeloma), Ganvado reduced the risk of developing a SRE by 19% (p = 0.0168) and developing multiple SREs by 15% (p = 0.0479) compared with zoledronic acid with the median time to first SRE delayed by 6 months.
In a post hoc analysis of Study 20050244 (including solid tumours, excluding multiple myeloma), Ganvado reduced the risk of developing a SRE by 19% (p = 0.0168) and developing multiple SREs by 15% (p = 0.0479) compared with zoledronic acid with the median time to first SRE delayed by 6 months.
Disease progression and overall survival in advanced malignancies involving bone.
Disease progression was similar between Ganvado and zoledronic acid in all three studies and in the pre-specified analysis of all three-studies combined.
In all three studies overall survival was balanced between Ganvado and zoledronic acid in patients with advanced malignancies involving bone: patients with breast cancer (hazard ratio [95% CI] was 0.95 [0.81, 1.11]), patients with prostate cancer (hazard ratio [95% CI] was 1.03 [0.91, 1.17]), and patients with other solid tumours or multiple myeloma (hazard ratio [95% CI] was 0.95 [0.83, 1.08]). A post hoc analysis in Study 20050244 (patients with other solid tumours or multiple myeloma) examined overall survival for the three tumour types used for stratification (non-small cell lung cancer, multiple myeloma, and other). Overall survival was longer for Ganvado in non-small cell lung cancer (hazard ratio [95% CI] of 0.79 [0.65, 0.95]; n = 702) and longer for zoledronic acid in multiple myeloma (hazard ratio [95% CI] of 2.26 [1.13, 4.50]; n = 180) and similar between the Ganvado and zoledronic acid groups in other tumour types (hazard ratio [95% CI] of 1.08 [0.90, 1.30]; n=894). This study did not control for prognostic factors and anti-neoplastic treatments. In a combined pre-specified analysis from all three studies, overall survival was similar between Ganvado and zoledronic acid (hazard ratio [95% CI] of 0.99 [0.91, 1.07]).
Clinical efficacy in patients with multiple myeloma. Ganvado was evaluated in an international, randomised (1:1), double-blind, active-controlled study comparing Ganvado with zoledronic acid in patients with newly diagnosed multiple myeloma (Study 20090482).
In this study, 1718 multiple myeloma patients with at least 1 bone lesion were randomised to receive 120 mg Ganvado subcutaneously every 4 weeks or 4 mg zoledronic acid intravenously (IV) every 4 weeks (dose adjusted for renal impairment and patients with creatinine clearance less than 30 mL/min were excluded based on Zometa prescribing information). The primary outcome measure was demonstration of non-inferiority of time to first skeletal-related event (SRE) as compared to zoledronic acid. Secondary outcome measures included superiority of time to first SRE, superiority of time to first and subsequent SRE, and overall survival. An SRE was defined as any of the following: pathologic fracture (vertebral or non-vertebral), radiation therapy to bone (including the use of radioisotopes), surgery to bone, or spinal cord compression.
In this study, randomisation was stratified by intent to undergo autologous peripheral blood stem cell (PBSC) transplantation (yes or no), the anti-myeloma agent being utilised/planned to be utilised in first-line therapy [novel therapy-based or non-novel therapy-based (novel therapies include bortezomib, lenalidomide, or thalidomide)], stage at diagnosis (International Staging System I or II or III), previous SRE (yes or no), and region (Japan or other countries). Across both study arms, 54.5% of patients intended to undergo autologous PBSC transplantation, 95.8% of patients utilised/planned to utilise a novel anti-myeloma agent in first-line therapy, and 60.7% of patients had a previous SRE. The number of patients across both study arms with ISS stage I, stage II, and stage III at diagnosis were 32.4%, 38.2%, and 29.3%, respectively.
Median age was 63 years, 82.1% of patients were White, and 45.6% of patients were women. The median number of doses administered was 16 for Ganvado and 15 for zoledronic acid. In patients with newly diagnosed multiple myeloma, Ganvado was non-inferior to zoledronic acid in delaying the time to first SRE following randomisation (see Figure 2 and Table 3).
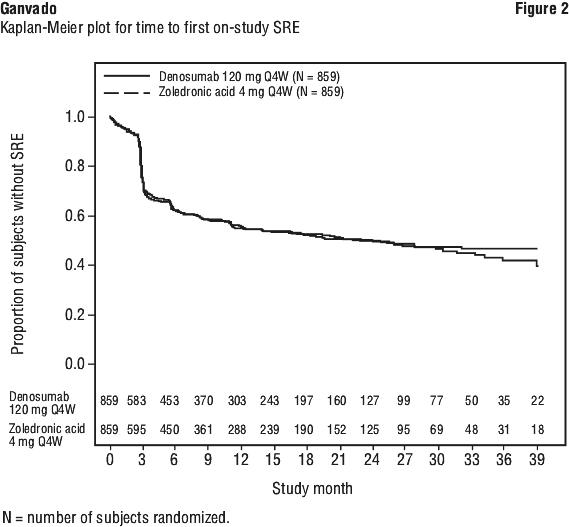
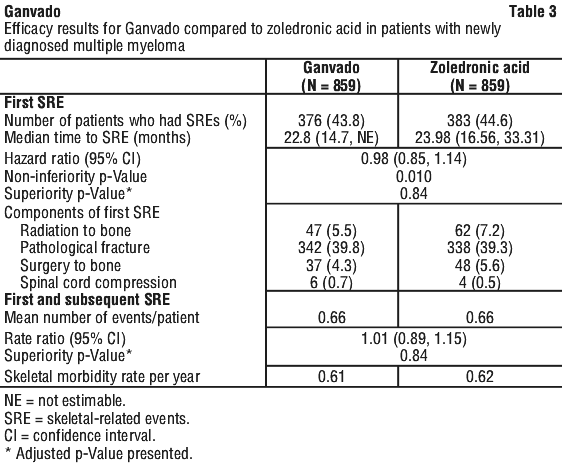
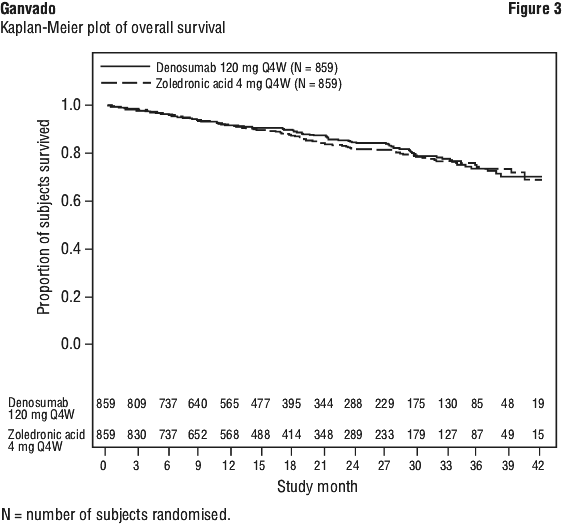
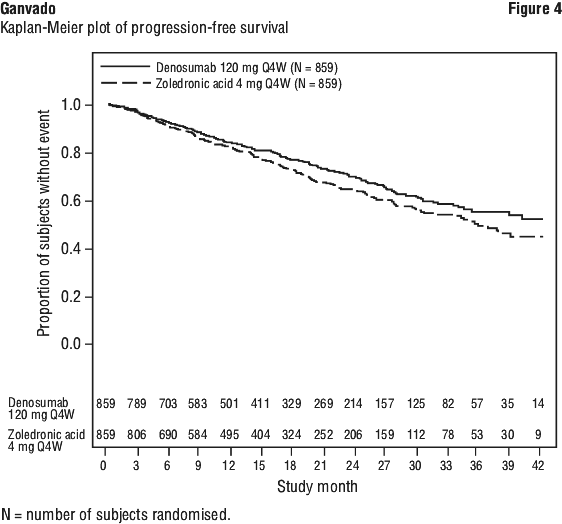
Overall survival and progression free survival in multiple myeloma.
The hazard ratio between Ganvado and zoledronic acid treatment groups and 95% CI for overall survival (OS) was 0.90 (0.70, 1.16) (see Figure 3). Progression-free survival (PFS) was assessed as an exploratory endpoint. Median PFS (95% CI) was 46.1 (34.3, not estimable) months for the Ganvado treatment group and 35.4 (30.2, not estimable) months for the zoledronic acid group (HR [95% CI] of 0.82 [0.68, 0.99]) (see Figure 4).
Effect on pain.
Levels of pain were examined using the Brief Pain Inventory - Short Form (BPI-SF) questionnaire as an exploratory endpoint.
For pain measures based on BPI-SF, the point estimate (95% CI) of the average AUC of the pain severity score, relative to baseline, was -0.72 (-0.92, -0.51) for Ganvado and -0.40 (-0.59, -0.20) for zoledronic acid, with a point estimate (95% CI) for the treatment difference of -0.32 (-0.60, -0.04) and p = 0.024.
Ganvado and zoledronic acid showed similar results in time to, and proportion by visit for ≥ 2-point decrease, ≥ 2-point increase, and > 4-point in worst pain score.
Other measures showed similar results between Ganvado and zoledronic acid, and results suggested that there were unlikely to be clinically significant differences between denosumab and zoledronic acid with regards to effects on pain.
Clinical efficacy in adults and skeletally mature adolescents with giant cell tumour of bone. The safety and efficacy of Ganvado was studied in two phase 2 open-label, single arm trials (Studies 20040215 and 20062004). The interim analysis, enrolled 305 patients with giant cell tumour of bone that was either unresectable or for which surgery would be associated with severe morbidity. Patients received 120 mg Ganvado subcutaneously every 4 weeks with a loading dose of 120 mg on days 8 and 15 of the initial 4-week treatment period.
Study 20040215 enrolled 37 adult patients with histologically confirmed unresectable or recurrent giant cell tumour of bone. The main outcome measure of the trial was response rate, defined as either at least 90% elimination of giant cells relative to baseline (or complete elimination of giant cells in cases where giant cells represent < 5% of tumour cells), or a lack of progression of the target lesion by radiographic measurements in cases where histopathology was not available.
Of the 35 patients included in the efficacy analysis, 85.7% (95% CI: 69.7, 95.2) had a treatment response to Ganvado. All 20 patients (100%) with histology assessments met response criteria. Of the remaining 15 patients, 10 (67%) met response criteria based on radiology data.
Study 20062004 interim analysis enrolled 282 adult or skeletally mature adolescents with giant cell tumour of bone. Patients were assigned to one of three cohorts: Cohort 1 included patients with surgically unsalvageable disease (e.g. sacral, spinal, or multiple lesions, including pulmonary metastases); Cohort 2 included patients with surgically salvageable disease whose planned surgery was associated with severe morbidity (e.g. joint resection, limb amputation, or hemipelvectomy); Cohort 3 included patients previously participating in 20040215 and rolled over into this study. The secondary outcome measures of the study were time to disease progression (based on investigator assessment) for Cohort 1 and proportion of patients without any surgery at month 6 for Cohort 2. Pain outcomes and investigator determined clinical benefit were also assessed.
In Cohort 1, median time to disease progression was not reached, as only 6 of the 169 treated patients (3.6%) had disease progression. In Cohort 2, Ganvado prolonged the time to surgery, reduced the morbidity of planned surgery, and reduced the proportion of patients undergoing surgery (see Table 4). Sixty-four of the 71 (90.1%; 95% CI: 80.7%, 95.9%) evaluable patients treated with Ganvado had not undergone surgery by month 6. Overall, of 100 patients for whom surgery was planned, 74 patients (74%) had no surgery performed, and 16 patients (16%) underwent a less morbid surgical procedure from that planned at baseline (see Table 4).
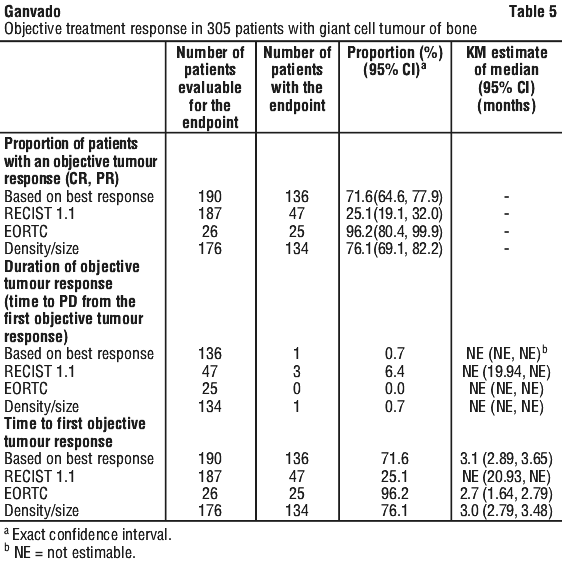
A retrospective independent review of radiographic imaging data was performed for patients enrolled in 20040215 and 20062004. Of the 305 patients enrolled in these studies, 190 had at least 1 evaluable timepoint response and were included in the analysis (see Table 5).
Patients were evaluated by the following response criteria to determine objective tumour response:
Modified Response Evaluation Criteria in Solid Tumours (RECIST 1.1) to evaluate tumour burden based on computed tomography (CT)/magnetic resonance imaging (MRI);
Modified European Organisation for Research and Treatment of Cancer (EORTC) criteria to evaluate metabolic response using fluorodeoxyglucose positron emission tomography (FDG-PET);
Modified Inverse Choi criteria to evaluate tumour size and density using Hounsfield units based on CT/MRI (Density/Size).
Ganvado achieved objective tumour responses in 136 of these 190 patients (71.6%; 95% CI: 64.6, 77.9) (see Table 5). The median time to response was 3.1 months (95% CI: 2.89, 3.65). The median duration of response was not estimable, as few patients experienced disease progression, with a median follow-up of 13.4 months. Efficacy results in skeletally mature adolescents appeared to be similar to those observed in adults.
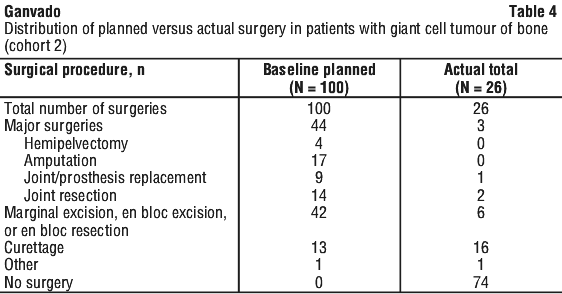
Effect on pain.
In Study 20062004 interim analysis, Cohorts 1 and 2 combined, a clinically meaningful reduction in worst pain (i.e. ≥ 2-point decrease from baseline) was reported for 31.4% of patients at risk (i.e. those who had a worst pain score of ≥ 2 at baseline) within 1 week of treatment, and ≥ 50% at week 5. These pain improvements were maintained at all subsequent evaluations. In a post-hoc analysis, at least half of evaluable patients had a ≥ 30% reduction in worst pain score from baseline at all post-baseline time points beginning at week 9. Overall, pain improvement and clinical benefit did not correlate with objective tumour response.
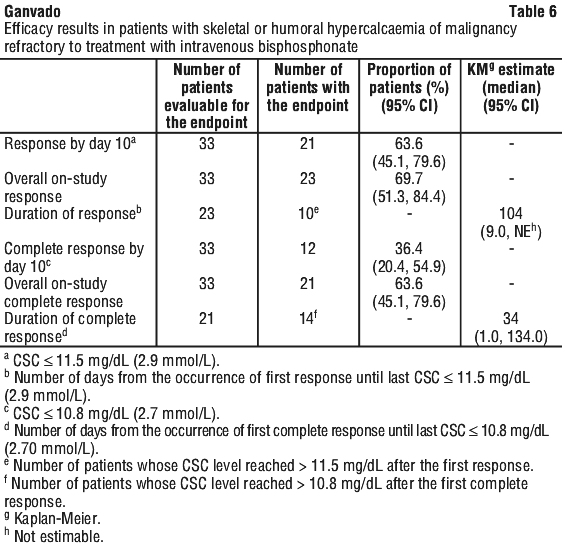
Clinical efficacy in treatment of hypercalcaemia of malignancy. The safety and efficacy of Ganvado was studied in a phase 2 open-label, single-arm trial (Study 20070315) that enrolled 33 patients with hypercalcaemia of malignancy (with or without bone metastases) refractory to treatment with intravenous bisphosphonate. In this study, refractory hypercalcaemia of malignancy was defined as an albumin-corrected serum calcium (CSC) of > 12.5 mg/dL (3.1 mmol/L) despite treatment with intravenous bisphosphonate in the last 7-30 days.
Twenty-six (79%) patients had advanced solid tumours and 7 (21%) patients had advanced hematologic malignancies. Twenty-five patients (76%) had poor performance status (Eastern Cooperative Oncology Group [ECOG] ≥ 2) at baseline. Metastatic disease was present in 30 (91%) patients and metastatic bone disease in 13 (39%) patients at baseline. Three (9%) patients had non-metastatic disease, 2 with myeloma and 1 with non-Hodgkin's lymphoma. Nineteen patients (58%) reported symptoms related to hypercalcaemia of malignancy at baseline. At the time of enrollment, the median serum calcium level was 13.7 mg/dL (3.42 mmol/L).
The primary endpoint was the proportion of patients achieving a response, defined as CSC ≤ 11.5mg/dL (2.9 mmol/L), within 10 days after Ganvado administration. The secondary objectives were to determine the duration of the treatment effect, the time to response, the time to relapse/nonresponse and to evaluate changes in CSC level from baseline.
Patients received Ganvado subcutaneously every 4 weeks with additional 120 mg doses on days 8 and 15 of the first month of therapy.
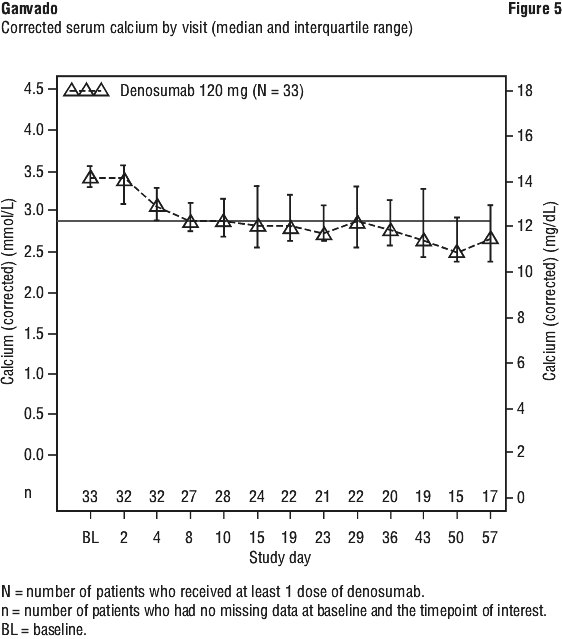
Ganvado was associated with rapid and sustained decreases in serum calcium in the majority of patients including those with or without bone metastases (see Figure 5 and Table 6).
Symptom improvement in patients with refractory hypercalcaemia of malignancy.
In Study 20070315, data regarding hypercalcaemia of malignancy symptoms were collected on a dedicated case report form. In the study population, a total of 48 hypercalcaemia of malignancy symptoms were reported in 19 patients at baseline. Each symptom status was based on the best status by study day 10.
8 (42%) patients reported resolution of at least 1 symptom.
4 (21%) patients reported resolution of all symptoms.
15 (31%) of the symptoms present at baseline resolved.
5 (10%) of the symptoms improved.
2 (4%) of the symptoms got worse.
26 (54%) of the symptoms remained stable.
Nine patients reported a total of 12 hypercalcaemia of malignancy symptoms of cognitive impairment at baseline. Each symptom status was based on the best status by study day 10.
5 (56%) of patients reported resolution of at least 1 symptom of cognitive impairment.
4 (44%) of patients reported resolution of all symptoms of cognitive impairment.
7 (58%) of the symptoms of cognitive impairment present at baseline resolved.
1 (8%) cognitive impairment symptom worsened.
4 (33%) of the cognitive impairment symptoms remained stable.
5.2 Pharmacokinetic Properties
Absorption.
Following subcutaneous administration, bioavailability was 62%.
Distribution.
Denosumab displayed non-linear pharmacokinetics with dose over a wide dose range, but approximately dose-proportional increases in exposure for doses of 60 mg (or 1 mg/kg) and higher.
In patients with advanced cancer who received multiple doses of 120 mg every 4 weeks (Q4W) an approximate 2-fold accumulation in serum denosumab concentrations was observed and steady-state was achieved by 6 months, consistent with time-independent pharmacokinetics.
At steady-state, the mean serum trough concentration was 20.6 microgram/mL (range: 0.456 to 56.9 microgram/mL). In patients with multiple myeloma who received 120 mg every 4 weeks, median trough levels varied by less than 8% between months 6 and 12.
In patients with giant cell tumour of bone who received 120 mg every 4 weeks with a loading dose on days 8 and 15, steady-state levels were achieved within the first month of treatment. Between weeks 9 and 49, median trough levels varied by less than 9%.
Metabolism.
Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin and is unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Excretion.
In patients with advanced cancer who discontinued doses of 120 mg every 4 weeks, the mean half-life was 28 days (range: 14 to 55 days).
Special populations.
A population pharmacokinetic analysis showed no notable difference in pharmacokinetics with age (18 to 87 years), race, body weight (36 to 174 kg), or across patients with solid tumours, multiple myeloma, and giant cell tumour of bone. The pharmacokinetics and pharmacodynamics of denosumab were similar in patients transitioning from IV bisphosphonate therapy.
Elderly.
The pharmacokinetics of denosumab were not affected by age (18 to 87 years).
Paediatric.
The pharmacokinetic profile has not been assessed in those < 18 years.
Impaired hepatic function.
The pharmacokinetic profile has not been assessed in patients with impaired hepatic function.
Impaired renal function.
In studies of denosumab (60 mg, N = 55 and 120 mg, N = 32) in patients without advanced malignancies but with varying degrees of renal function, including patients on dialysis, the degree of renal impairment had no effect on the pharmacokinetics and pharmacodynamics of denosumab. Dose adjustment for renal impairment is not necessary.
Immunogenicity.
Denosumab pharmacokinetics and pharmacodynamics were not affected by the formation of binding antibodies to denosumab and were similar in men and women.
In clinical studies, no neutralising antibodies for denosumab have been observed in advanced cancer patients or giant cell tumour of the bone patients. Using a sensitive immunoassay, < 1% of patients treated with denosumab for up to 3 years tested positive for non-neutralising binding antibodies with no evidence of altered pharmacokinetics, toxicity, or clinical response.
5.3 Preclinical Safety Data
Genotoxicity.
The genotoxic potential of denosumab has not been evaluated. Denosumab is a recombinant protein comprised entirely of naturally occurring amino acids and contains no inorganic or synthetic organic linkers or other non-protein portions. Therefore, it is unlikely that denosumab or any of its derived fragments would react with DNA or other chromosomal material.
Carcinogenicity.
The carcinogenic potential of denosumab has not been evaluated in long-term animal studies. In view of the mechanism of action of denosumab, it is unlikely that the molecule would be capable of inducing tumour development or proliferation.6 Pharmaceutical Particulars
6.1 List of Excipients
Each single-use vial of Ganvado contains 78.1 mg sorbitol, 1.8 mg glacial acetic acid, 0.17 mg polysorbate 20 and sodium hydroxide for adjusting to pH 5.2 in water for injections.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
It is recommended to store Ganvado vials in a refrigerator at 2°C to 8°C in the original carton. Do not freeze. Protect from direct light. Do not excessively shake the vial. Do not expose to temperatures above 25°C.
If removed from the refrigerator, Ganvado should be kept at room temperature (up to 25°C) in the original container and must be used within 30 days.
6.5 Nature and Contents of Container
Ganvado is supplied in a glass vial.
Pack size: one or four* vials.
*Not marketed in Australia.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.

CAS number.
615258-40-7.7 Medicine Schedule (Poisons Standard)
S4 Prescription Medicine.
Summary Table of Changes
