What is in this leaflet
This leaflet answers some common questions about Glivec.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine. You can also download the most up to date leaflet from www.novartis.com.au. Those updates may contain important information about the medicine and its use of which you should be aware.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will provide.
If you have any concerns about this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Glivec is used for
Glivec is used to treat adults and children/adolescents who have chronic myeloid leukaemia (CML) and acute lymphoblastic leukaemia with Philadelphia chromosome positive (Ph-positive ALL).
CML and ALL are types of leukaemia in which an abnormal chromosome produces an enzyme that leads to uncontrolled growth of white blood cells. Glivec kills the abnormal cells while leaving normal cells alone.
Glivec is also used to treat adults for:
- myelodysplastic / myeloproliferative diseases (MDS/MPD).
These are a group of blood diseases in which some blood cells start growing out of control. - Aggressive systemic mastocytosis (ASM).
It is a cancer in which certain blood cells, called "mast" cells, grow out of control. - Hypereosinophilic syndrome (HES) and or chronic eosinophilic leukaemia (CEL).
These are blood diseases in which some blood cells, named "eosinophils", start growing out of control. - gastro-intestinal stromal tumours (GIST).
This is a type of cancer of the stomach and bowels. This cancer affects the tissue that surrounds the stomach and bowels and the cells grow uncontrollably. - dermatofibrosarcoma protuberans (DFSP).
DFSP is a cancer of the tissue beneath the skin in which some cells start growing out of control.
Glivec works by slowing the growth of abnormal cells. Glivec kills the abnormal cells while leaving normal cells alone.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another purpose.
Glivec is only available with a doctor's prescription. It is not addictive.
There is not enough information to recommend the use of Glivec in children under 3 years of age for most uses. For use in CML, there is no experience with the use of Glivec in children below 2 years of age. For acute lymphoblastic leukaemia with Philadelphia chromosome positive (Ph-positive ALL), there is no experience with the use of Glivec in children below 1 year of age.
Before you take Glivec
When you must not take it
Do not take Glivec if you have ever had an allergic reaction to imatinib (the active ingredient) or to any of the other ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
Do not take Glivec after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. In that case, return the medicine to your pharmacist.
Before you start to take it
Tell your doctor if you are pregnant or planning to become pregnant. This medicine may be harmful to your unborn baby. If it is necessary for you to take it during pregnancy, your doctor will discuss with you the risks and benefits involved.
Tell your doctor if you are breast-feeding. It is not known if the active ingredient, imatinib, passes into the breast milk. Because this medicine could affect your baby, breast-feeding is not recommended.
Tell your doctor if you have ever had any of the following medical problems or procedures:
- kidney or liver problems
- problems with your heart
- you have had your thyroid gland removed
- hepatitis B infection. As during treatment with Glivec, hepatitis B (an infection of the liver) may become active again.
Your doctor may want to take special precautions in that case.
Taking other medicines
Tell your doctor if you are taking any other medicines, including medicines that you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines and Glivec may interfere with each other. These include many medicines that are eliminated from the body through the liver:
- St. John's wort, a herbal medicine found in many products that you can buy without a prescription
- paracetamol, a medicine found in many common pain relievers and cold remedies (e.g. Panadol®, Panadeine®, Codral®, Tylenol®) which are known to be associated with liver toxicity.
- A patient, who was taking paracetamol regularly for fever, died of acute liver failure. Although the cause is currently unknown, special caution should be exercised when using paracetamol and Glivec.
- antibiotic medicines such as rifampicin, ketoconazole, erythromycin, clarithromycin, itraconazole
- antiviral medicines used to treat HIV/AIDS
- dexamethasone, a steroid medicine
- medicines for high cholesterol, such as simvastatin
- medicines used to treat epilepsy, such as phenytoin, carbamazepine, phenobarbitone
- warfarin, a medicine used to prevent blood clots
- some medicines used to treat mental disorders and depression
- some medicines used to treat high blood pressure and heart problems
- cyclosporin
You may need to take different amounts of these medicines or you may need to take different medicines. Your doctor and pharmacist have more information.
If you have not told your doctor about any of these things, tell him/her before you start taking this medicine.
How to take Glivec
Follow all directions given to you by your doctor and pharmacist carefully. These instructions may differ from the information contained in this leaflet.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
For CML, the usual dose for an adult is 400 to 600 mg each day and the maximum dose is 800 mg each day. The dose depends on what stage of CML you have. For Ph-positive ALL the usual dose is 600 mg each day. For children treated with CML and Ph-positive ALL, the dose depends on the size of the child.
For MDS/MPD, the starting dose is 400 mg.
For ASM and HES/CEL, the usual starting dose is 400 mg. For some patients the starting dose may be 100 mg.
For GIST, the usual dose is 400 mg or 600 mg each day.
For DFSP, the starting dose is 800 mg per day.
Daily dose of 400 mg should be taken as one tablet of 400 mg once a day.
Daily dose of 600 mg should be taken as either:
- six tablets of 100 mg or
- one tablet of 400 mg plus half a 400 mg tablet once a day.
Daily dose of 800 mg should be taken as 400 mg twice a day, in the morning and in the evening.
Your doctor may direct you to take a higher or lower dose, or stop treatment if needed depending on your response to Glivec.
Glivec is usually taken as a single dose each day. However, your doctor may want you to take them in two doses, one in the morning and one in the evening.
How to take it
Take the medicine with a large glass of water and food. This will help to avoid irritating the lining of your oesophagus (food pipe) and stomach.
If you are unable to swallow the tablets:
- Put the required tablet(s) in a glass of water or apple juice (approximately 50 ml for a 100 mg tablet and 200 ml for a 400 mg tablet).
- Stir with a spoon to completely disintegrate the tablet(s).
- Immediately drink the whole contents of the glass.
For the best effect, take the medicine at about the same time each day. Taking them at the same time each day will help you to remember to take them.
How long to take it
Continue taking Glivec every day for as long as your doctor prescribes.
Your doctor will keep a close check on you to make sure you are still benefiting from treatment.
If you forget to take it
Take the missed dose as soon as you remember, then continue with your normal schedule.
Do not take a double dose to make up for the one that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much (Overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone number 13 11 26), or go to Accident and Emergency at your nearest hospital if you think that you or anyone else may have taken too much Glivec. Do this even if there are no signs of discomfort or poisoning. Keep the telephone numbers for these places handy.
While you are taking Glivec
Things you must do
Make sure you follow your doctor's instructions carefully and keep all appointments.
You will need regular follow-up to make sure the treatment is working. Regular blood tests, weight checks and urine tests can also find side effects before they become serious.
Some children and adolescents taking Glivec may have slower than normal growth. Growth will be monitored at regular visits by your doctor.
Make sure you use a method of contraception to prevent pregnancy during treatment with Glivec and for 15 days after ending treatment. Tell your doctor immediately if you become pregnant while you are taking this medicine.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Glivec.
Tell any other doctor, dentist or pharmacist who treats you that you are taking Glivec.
Things you must not do
Do not give this medicine to anyone else even if their condition seems to be the same as yours.
Do not use it to treat any other complaints unless your doctor tells you to.
Things to be careful of
Avoid drinking grapefruit juice while you are being treated with Glivec. Grapefruit juice may interact with Glivec and affect how your body uses this medicine.
If you need to take something to treat a headache, cold or other minor aches and pains, try to avoid taking medicines containing paracetamol (e.g. Panadol®, Panadeine®, Codral®, Tylenol®). Ask your pharmacist to suggest an alternative medicine.
Be careful driving, operating machinery or doing jobs that require you to be alert until you know how Glivec affects you. This medicine may cause dizziness, light-headedness or drowsiness in some people. Make sure you know how you react to it before you drive a car, operate machinery or do anything that could be dangerous.
When you are outdoors, wear protective clothing and use at least a 15+ sunscreen. Do not use sunlamps or tanning beds. This medicine may cause your skin to be much more sensitive to sunlight than it normally is.
Exposure to sunlight may cause a skin rash, itching, redness or severe sunburn. If your skin does appear to be burning, tell your doctor.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Glivec.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Do not be alarmed by these lists of possible side effects. You may not experience any of them. Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following side effects and they worry you:
- swelling of fingers, eyelids, face or lower legs due to fluid build up (see your doctor immediately if fluid build up is severe)
- indigestion, upset stomach, wind, feeling of bloating
- nausea (feeling sick) or vomiting
- diarrhoea
- constipation
- dry mouth
- swelling, aching, cramping or stiffness in joints or muscles
- musculoskeletal pain after stopping Glivec (including muscle pain, limb pain, joint pain, bone pain and back pain)
- pain in the bones or along veins
- headache
- dizziness, light-headedness or vertigo (spinning sensation)
- tiredness, weakness, feeling generally unwell
- numbness, coldness or tingling in fingers and toes
- difficulty sleeping, feeling anxious, depressed, confused or forgetful
- change in sense of taste
- rash, eczema, itching, dry skin, darkening or lightening of skin
- symptoms of sunburn (such as redness, itching, swelling or blistering of the skin) which happens more quickly than normal
- irritated, red, runny or itchy eyes, blurred vision
- ringing in the ears
- changes in appetite and weight
- hair loss
- sweating during the night
- throat pain
- cough or cold symptoms
- loss of interest in sex, problems with sexual function
- breast enlargement, nipple pain, painful periods
- reddening and/or swelling on the palms of the hands and soles of the feet which may be accompanied by tingling sensation and burning pain
- slowing of growth in children and adolescents
Tell your doctor immediately if you get any of the following side effects:
- severe allergic reaction that can result in difficulty breathing, dizziness.
- rapid weight gain, swelling of the extremities (calves, ankles), generalised swelling such as swelling of the face (signs of water retention)
- weakness, spontaneous bleeding or bruising, frequent infections with signs such as fever, chills, swollen glands, sore throat or mouth ulcers (signs of low level of blood cells)
- pale skin, tiredness, breathlessness, dark urine (signs of break down of red blood cells).
- pain and having difficulty walking
- cough, difficult or painful breathing, wheezing, pain in chest when breathing (signs of lung infections/disorders).
- muscle weakness, muscle spasms, abnormal heart rhythm (signs of changes in level of potassium in the blood).
- muscle spasms, fever, red-brown urine, kidney disorders, pain or weakness in muscles (signs of muscle disorders)
- severe abdominal pain, vomiting blood, black or bloody stools, swelling of the abdomen/fluid within the abdomen, constipation, stomach pain (signs of gastrointestinal disorders)
- thirst, weight loss and severely decreased urine output (signs of low intake of drinks/fluids)
- nosebleeds or any other unusual bleeding
- vision impairment, blurred vision, blood in eye
- nausea, loss of appetite, dark-coloured urine or yellowing of your skin or eyes (signs of liver disorders).
- Changes in urine or blood in urine, pain in the kidney area, tiredness, loss of appetite, nausea/ vomiting, lack of concentration, headache, cramping, itching (signs of kidney disorders)
- nausea, diarrhoea, vomiting, abdominal pain, fever (signs of inflammatory bowel disease).
- severe rash, red skin, blistering of the lips, eyes, skin or mouth, skin peeling, fever, red raised or purple skin patches, itching, burning, pustular eruption (signs of skin disorder).
- severe skin rash, itching, hives, blisters or peeling skin, which may be accompanied by fever, chills, headache, swollen glands, stomach pain or aching joints and muscles
- inflammation of the skin caused by an infection (sign of cellulitis)
- blood in the urine
- severe headache, weakness or paralysis of limbs or face, difficulty speaking, sudden loss of consciousness (signs of nervous system disorder such as bleeding or swelling in the brain)
- seizures (fits)
- swelling and pain in one part of the body (signs of clots in blood vessels)
- crushing chest pain, fever, tiredness, irregular heart beat (signs of heart disorders such as heart attack, angina).
- muscle weakness, muscle spasms, abnormal heart rhythm (signs of changes in level of potassium in the blood)
- pelvic pain sometimes accompanied by nausea and vomiting, unexpected vaginal bleeding, (signs of gynaecological disorder)
- nausea, shortness of breath, irregular heartbeat, clouding of urine, tiredness and/or joint discomfort associated with abnormal laboratory (such as high potassium, uric acid, and phosphorous levels and low calcium levels in the blood)
- severe headache, dizziness, blurred vision (signs of increased pressure inside skull)
- fever, skin rash, joint pain and swelling as well as tiredness, loss of appetite, nausea, jaundice (yellowing of the skin), pain in the upper right abdomen, pale stools and dark urine (potential signs of hepatitis B reactivation).
- Fatigue, dizziness, shortness of breath, bruises, gum/nose bleeds, minor cuts that bleed a lot, confusion, sleepiness, seizures, decreased urine, swollen legs, fever (potential signs of thrombotic microangiopathy).
The above side effects may be serious. You may need urgent medical attention.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed here or not yet known may happen in some people. Some of these side effects can only be found by laboratory testing.
After taking Glivec
Storage
- Keep your medicine in the original container until it is time to take it.
- Store it in a cool dry place.
- Do not store Glivec or any other medicine in the bathroom or near a sink.
- Do not leave it in the car or on window sills.
Keep the medicine where young children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine you have left over.
Product description
What it looks like
Tablets
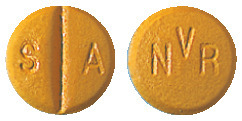
Glivec 100 mg tablet is a round, very dark yellow to brownish orange tablet with NVR on one side and SA and score-line on the other side; packs of 60 tablets.
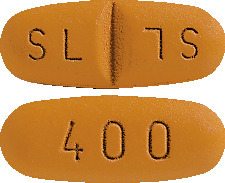
Glivec 400 mg tablet is an oval, very dark yellow to brownish orange tablet with 400 on one side and score on the other side and SL on each side of the score; packs of 30 tablets.
Ingredients
Glivec tablets contain 100 mg or 400 mg of imatinib. Glivec tablets also contain:
- cellulose-microcrystalline
- crospovidone
- hypromellose
- silica colloidal anhydrous
- magnesium stearate
- iron oxide yellow CI77492 (E172)
- iron oxide red CI77491 (E172)
- macrogol 4000
- talc
Sponsor
Glivec is supplied in Australia by:
Novartis Pharmaceuticals Australia Pty Limited
(ABN 18 004 244 160)
54 Waterloo Road
Macquarie Park NSW 2113
Telephone 1 800 671 203
Web site: www.novartis.com.au
® = Registered Trademark
This leaflet was prepared in September 2021.
Australian Registration Number.
Glivec 100 mg Tablets AUST R 94216
Glivec 400 mg Tablets AUST R 94217
Glivec 100 mg Capsules AUST R 78442 *
*Capsule packs are not available in Australia
(gli230921c.doc) based on PI (gli230921i.doc)
Published by MIMS November 2021





 The adjuvant trial SSG XVIII/AIO compared 1 year and 3 years Glivec treatment. Table 7 shows adverse events, regardless of relationship to study drug, that were reported in at least 5% of patients treated with Glivec.
The adjuvant trial SSG XVIII/AIO compared 1 year and 3 years Glivec treatment. Table 7 shows adverse events, regardless of relationship to study drug, that were reported in at least 5% of patients treated with Glivec. Adverse events with imatinib observed in trials in Ph+ ALL, MDS/MPD, SM, HES/CEL and DFSP were generally consistent with those seen in CML and GIST studies.
Adverse events with imatinib observed in trials in Ph+ ALL, MDS/MPD, SM, HES/CEL and DFSP were generally consistent with those seen in CML and GIST studies.


 Laboratory abnormalities with imatinib observed in trials in Ph+ ALL, MDS/MPD, SM, HES/CEL and DFSP were generally consistent with those seen in CML and GIST studies.
Laboratory abnormalities with imatinib observed in trials in Ph+ ALL, MDS/MPD, SM, HES/CEL and DFSP were generally consistent with those seen in CML and GIST studies. Rates of complete haematological response, major cytogenetic response and complete cytogenetic response on first-line treatment were estimated using the Kaplan-Meier approach, for which non-responses were censored at the date of last examination. Using this approach the estimated cumulative response rates for first-line treatment with Glivec are shown in Table 13.
Rates of complete haematological response, major cytogenetic response and complete cytogenetic response on first-line treatment were estimated using the Kaplan-Meier approach, for which non-responses were censored at the date of last examination. Using this approach the estimated cumulative response rates for first-line treatment with Glivec are shown in Table 13. For analysis of long-term outcomes patients randomised to receive Glivec were compared with patients randomised to receive IFN. Patients who crossed over prior to progression were not censored at the time of crossover, and events that occurred in these patients following crossover were attributed to the original randomised treatment.
For analysis of long-term outcomes patients randomised to receive Glivec were compared with patients randomised to receive IFN. Patients who crossed over prior to progression were not censored at the time of crossover, and events that occurred in these patients following crossover were attributed to the original randomised treatment.
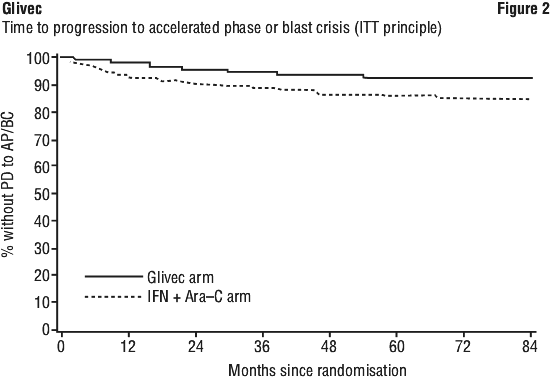 A total of 71 (12.8%) and 85 (15.4%) patients died in the Glivec and IFN + Ara-C groups, respectively. At 84 months the estimated overall survival is 86.4% (83, 90) vs. 83.3% (80, 87) in the randomised Glivec and the IFN + Ara-C groups, respectively (p = 0.073, log-rank test).
A total of 71 (12.8%) and 85 (15.4%) patients died in the Glivec and IFN + Ara-C groups, respectively. At 84 months the estimated overall survival is 86.4% (83, 90) vs. 83.3% (80, 87) in the randomised Glivec and the IFN + Ara-C groups, respectively (p = 0.073, log-rank test).
 Efficacy results were similar in men and women and in patients younger and older than age 65. Responses were seen in black patients, but there were too few black patients to allow a quantitative comparison.
Efficacy results were similar in men and women and in patients younger and older than age 65. Responses were seen in black patients, but there were too few black patients to allow a quantitative comparison. In study AJP01, imatinib (600 mg/day on days 8-63 of induction chemotherapy, and on days 1 - 28 of each chemotherapy cycle during consolidation and maintenance) was integrated into a chemotherapy regimen in 80 patients with de novo Ph+ ALL. Results are summarized in Table 17.
In study AJP01, imatinib (600 mg/day on days 8-63 of induction chemotherapy, and on days 1 - 28 of each chemotherapy cycle during consolidation and maintenance) was integrated into a chemotherapy regimen in 80 patients with de novo Ph+ ALL. Results are summarized in Table 17. Analysis of event-free survival and overall survival also indicated superiority of the imatinib-containing regimen (p < 0.0001 for both).
Analysis of event-free survival and overall survival also indicated superiority of the imatinib-containing regimen (p < 0.0001 for both).

 Glivec has not been shown to be effective in patients with less aggressive forms of systemic mastocytosis. Glivec is not recommended for use in patients with cutaneous mastocytosis, indolent systemic mastocytosis (smoldering SM or isolated bone marrow mastocytosis), SM with an associated clonal haematological non-mast cell lineage disease, mast cell leukaemia, mast cell sarcoma or extracutaneous mastocytoma. In vitro, cell lines and patient-derived mast cells harbouring the KIT D816V mutation were resistant to imatinib and the effectiveness of Glivec in the treatment of patients with SM who have the D816V mutation remains controversial.
Glivec has not been shown to be effective in patients with less aggressive forms of systemic mastocytosis. Glivec is not recommended for use in patients with cutaneous mastocytosis, indolent systemic mastocytosis (smoldering SM or isolated bone marrow mastocytosis), SM with an associated clonal haematological non-mast cell lineage disease, mast cell leukaemia, mast cell sarcoma or extracutaneous mastocytoma. In vitro, cell lines and patient-derived mast cells harbouring the KIT D816V mutation were resistant to imatinib and the effectiveness of Glivec in the treatment of patients with SM who have the D816V mutation remains controversial. Additionally, improvements in symptomatology and other organ dysfunction abnormalities were reported by the investigators in the case reports. Improvements were reported in cardiac, nervous, skin/subcutaneous tissue, respiratory/thoracic/mediastinal, musculoskeletal/connective tissue/vascular, and gastrointestinal organ systems.
Additionally, improvements in symptomatology and other organ dysfunction abnormalities were reported by the investigators in the case reports. Improvements were reported in cardiac, nervous, skin/subcutaneous tissue, respiratory/thoracic/mediastinal, musculoskeletal/connective tissue/vascular, and gastrointestinal organ systems.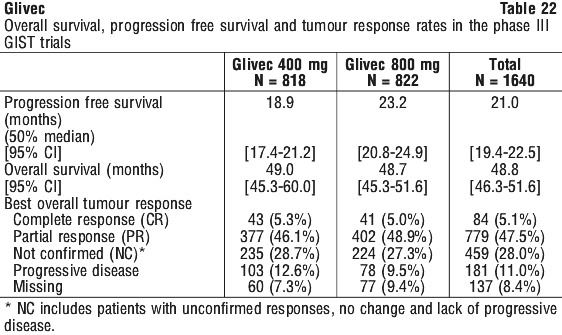 Median follow up for the combined studies was 37.5 months (25th - 75th percentile 19 to 46 months). There was a statistically significant improvement in PFS in the 800 mg treatment group (23.2 months [95% CI, 20.8 to 24.9]) compared to the 400 mg treatment group (18.9 months [95% CI, 17.4 to 21.2]) (p = 0.03). However, there were no observed differences in overall survival between the treatment groups (p = 0.98). The estimated overall PFS for all 1640 patients in these Phase III studies was 21 months [95% CI 19.4 to 22.5] and the estimated OS of 48.8 months [95% CI 46.3 to 51.6]. 5.1% of patients achieved a confirmed complete response and 47.5% achieved a partial response. Treatment at either dose level was generally well tolerated and overall 5.4% of patients withdrew due to toxicity.
Median follow up for the combined studies was 37.5 months (25th - 75th percentile 19 to 46 months). There was a statistically significant improvement in PFS in the 800 mg treatment group (23.2 months [95% CI, 20.8 to 24.9]) compared to the 400 mg treatment group (18.9 months [95% CI, 17.4 to 21.2]) (p = 0.03). However, there were no observed differences in overall survival between the treatment groups (p = 0.98). The estimated overall PFS for all 1640 patients in these Phase III studies was 21 months [95% CI 19.4 to 22.5] and the estimated OS of 48.8 months [95% CI 46.3 to 51.6]. 5.1% of patients achieved a confirmed complete response and 47.5% achieved a partial response. Treatment at either dose level was generally well tolerated and overall 5.4% of patients withdrew due to toxicity. A statistically significant difference in response rates between the two dose groups was not demonstrated. A significant number of patients who had stable disease at the time of the interim analysis achieved a partial response with longer treatment (median follow-up 31 months). Median time to response was 13 weeks (95% C.I. 12 to 23). Median time to treatment failure in responders was 122 weeks (95% C.I. 106 to 147), while in the overall study population it was 84 weeks (95% C.I. 71 to 109). The median overall survival has not been reached. The Kaplan-Meier estimate for survival after 36-month follow-up is 68% [Figure 3]. Additionally, there is no difference in survival between patients achieving stable disease and partial response [Figure 4].
A statistically significant difference in response rates between the two dose groups was not demonstrated. A significant number of patients who had stable disease at the time of the interim analysis achieved a partial response with longer treatment (median follow-up 31 months). Median time to response was 13 weeks (95% C.I. 12 to 23). Median time to treatment failure in responders was 122 weeks (95% C.I. 106 to 147), while in the overall study population it was 84 weeks (95% C.I. 71 to 109). The median overall survival has not been reached. The Kaplan-Meier estimate for survival after 36-month follow-up is 68% [Figure 3]. Additionally, there is no difference in survival between patients achieving stable disease and partial response [Figure 4].




 Twelve of these 18 patients either achieved a complete response (7 patients) or were made disease free by surgery after a partial response (5 patients, including one child) for a total complete response rate of 67%. A further 3 patients achieved a partial response, for an overall response rate of 83%. Of the 8 patients with metastatic disease, five responded (62%), three of them completely (37%). The median duration of therapy in study B2225 was 6.2 months, with a maximum duration of 24.3 months, while in the published literature it ranged between 4 weeks and more than 20 months.
Twelve of these 18 patients either achieved a complete response (7 patients) or were made disease free by surgery after a partial response (5 patients, including one child) for a total complete response rate of 67%. A further 3 patients achieved a partial response, for an overall response rate of 83%. Of the 8 patients with metastatic disease, five responded (62%), three of them completely (37%). The median duration of therapy in study B2225 was 6.2 months, with a maximum duration of 24.3 months, while in the published literature it ranged between 4 weeks and more than 20 months. Imatinib and its metabolites are not excreted via the kidney to a significant extent. In a study of patients with varying degrees of renal dysfunction (mild, moderate and severe, see Table 27 for renal function classification), the mean exposure to imatinib (dose normalized AUC) increased 1.5- to 2-fold compared to patients with normal renal function, which corresponded to an elevated plasma level of AGP, a protein to which imatinib binds strongly. No correlation between imatinib exposure and the severity of renal deficiency was observed. In this study, 800 mg daily was safely used in patients with mild renal dysfunction and 600 mg daily was used in moderate renal dysfunction. The 800 mg dose was not tested in patients with moderate renal dysfunction due to the limited number of patients enrolled. Similarly, only 2 patients with severe renal dysfunction were enrolled at the low (100 mg) dose, and no higher doses were tested. No patients on haemodialysis were enrolled in the study. Literature data showed that a daily dose of 400 mg was well tolerated in a patient with end-stage renal disease on haemodialysis. The PK plasma exposure in this patient fell within the range of values of imatinib and its metabolite CGP74588 observed in patients with normal renal function. Dialysis was not found to intervene with the plasma kinetics of imatinib. Since renal excretion represents a minor elimination pathway for imatinib, patients with severe renal insufficiency and on dialysis could receive treatment at the 400 mg starting dose. However, in these patients caution is recommended. The dose can be reduced if not tolerated, or increased for lack of efficacy (see Section 4.4 Special Warnings and Precautions for Use; Section 4.2 Dose and Method of Administration).
Imatinib and its metabolites are not excreted via the kidney to a significant extent. In a study of patients with varying degrees of renal dysfunction (mild, moderate and severe, see Table 27 for renal function classification), the mean exposure to imatinib (dose normalized AUC) increased 1.5- to 2-fold compared to patients with normal renal function, which corresponded to an elevated plasma level of AGP, a protein to which imatinib binds strongly. No correlation between imatinib exposure and the severity of renal deficiency was observed. In this study, 800 mg daily was safely used in patients with mild renal dysfunction and 600 mg daily was used in moderate renal dysfunction. The 800 mg dose was not tested in patients with moderate renal dysfunction due to the limited number of patients enrolled. Similarly, only 2 patients with severe renal dysfunction were enrolled at the low (100 mg) dose, and no higher doses were tested. No patients on haemodialysis were enrolled in the study. Literature data showed that a daily dose of 400 mg was well tolerated in a patient with end-stage renal disease on haemodialysis. The PK plasma exposure in this patient fell within the range of values of imatinib and its metabolite CGP74588 observed in patients with normal renal function. Dialysis was not found to intervene with the plasma kinetics of imatinib. Since renal excretion represents a minor elimination pathway for imatinib, patients with severe renal insufficiency and on dialysis could receive treatment at the 400 mg starting dose. However, in these patients caution is recommended. The dose can be reduced if not tolerated, or increased for lack of efficacy (see Section 4.4 Special Warnings and Precautions for Use; Section 4.2 Dose and Method of Administration).
 Chemical name: 4-[(4-methyl-1-piperazinyl) methyl]-N-[4-methyl-3- [[4-(3-pyridinyl)- 2-pyrimidinyl] amino]-phenyl]benzamide methanesulfonate.
Chemical name: 4-[(4-methyl-1-piperazinyl) methyl]-N-[4-methyl-3- [[4-(3-pyridinyl)- 2-pyrimidinyl] amino]-phenyl]benzamide methanesulfonate.