1 Name of Medicine
Hepcludex (bulevirtide acetate).
2 Qualitative and Quantitative Composition
Each vial contains bulevirtide acetate equivalent to 2 mg bulevirtide.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Hepcludex (bulevirtide) powder for injection, 2 mg, available as a sterile, preservative free, white to off-white lyophilized powder that is to be reconstituted with 1 mL of Sterile Water for Injection prior to administration by subcutaneous injection. Following reconstitution, each vial contains 2 mg/mL of bulevirtide solution with a pH of approximately 9.0 and osmolality of approximately 300 mOsm/kg.
4.1 Therapeutic Indications
Hepcludex is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in adults with compensated liver disease.
4.2 Dose and Method of Administration
Adults.
The recommended dosage in adults is Hepcludex 2 mg once daily administered by subcutaneous injection.
The optimal treatment duration is unknown. Treatment should be continued as long as associated with clinical benefit.
Consideration to discontinue the treatment should be given in case of sustained (6 months) HBsAg seroconversion.
In all patients, manage the underlying hepatitis B virus (HBV) infection simultaneously as clinically appropriate.
Dose preparation and administration.
Healthcare professionals should train patients in the proper technique for reconstituting Hepcludex with Sterile Water for Injection and self-administering subcutaneous injections using a syringe. Bulevirtide solution should be inspected visually for particulate matter and discoloration prior to administration.
Instruct the patient or caregiver to read the Instructions For Use at the time they receive a prescription for Hepcludex and as needed for ongoing administration of Hepcludex. Emphasize the following instructions to the patient or caregiver:
Hepcludex must be stored in the refrigerator prior to preparation and administration.
Hepcludex needs to be reconstituted with Sterile Water for Injection prior to administration.
The Sterile Water for Injection and syringe and needles for preparation and injection are provided separately from Hepcludex; they should be stored out of the reach of children.
Hepcludex must be administered by subcutaneous injection. Do not administer by any other route.
Reconstitution instructions.
Aseptically reconstitute Hepcludex lyophilized powder by adding 1 mL of Sterile Water for Injection to the Hepcludex vial.
Carefully tap and then roll the vial between the hands to dissolve the powder. Complete dissolution might take up to 3 minutes.
Completely dissolved Hepcludex should be clear without foam. Do not shake. If the Hepcludex solution appears foamy, allow more time for the powder to dissolve.
If there are bubbles in the solution, gently tap the vial until they disappear.
If there are particles in the solution once the powder is (completely) dissolved (after approximately 5 minutes), do not use that vial of solution.
Use reconstituted product immediately. Do not refrigerate.
Administration instructions.
Administer by subcutaneous injection into the upper thigh or lower abdomen.
If a dose is missed, that dose should be taken as soon as possible on that day. However, if it is almost time for the next dose, skip the missed dose and go back to the regular dosing schedule. Do not double doses.
Change the injection site with each injection.
Important: Remind patients that it is important that they do not reuse the vials, syringe, needles or any remaining sterile water for injection. Hepcludex and auxiliary supplies are for Single Use Only. Throw away (dispose of) all components after use, including unused Sterile Water for Injection.
Disposal of medicinal product and auxiliary components.
In Australia, all used components/waste should be handled according to the local regulation.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Special populations.
Use in hepatic impairment.
No dosage adjustment of Hepcludex is required in patients with mild hepatic impairment (Child-Pugh A). The safety and efficacy of Hepcludex in patients with Child-Pugh B or C hepatic impairment or patients with decompensated liver disease have not been evaluated.
Use in renal impairment.
No dosage adjustment of Hepcludex is required in patients with mild renal impairment (creatinine clearance [CrCl] ≥ 60 and < 90 mL/min). The safety and efficacy of Hepcludex in patients with CrCl < 60 mL/min have not been evaluated.
Use in the elderly.
No data are available on which to make a dose recommendation for patients over the age of 65 years.
Paediatric use.
The safety and efficacy of Hepcludex in patients younger than 18 years of age have not been evaluated.4.3 Contraindications
None.
4.4 Special Warnings and Precautions for Use
Exacerbation of hepatitis after discontinuation of treatment.
Severe acute exacerbations of HDV and HBV infection may occur after Hepcludex is discontinued, especially in patients with cirrhosis, who may be at increased risk of more severe flares or progression to hepatic decompensation. Monitor hepatic function closely with both clinical and laboratory follow-up for at least six months in patients who discontinue Hepcludex. In certain circumstances, resumption of antiviral therapy may be warranted.4.5 Interactions with Other Medicines and Other Forms of Interactions
In a clinical pharmacokinetic drug interaction study in healthy volunteers, there was no significant effect of bulevirtide on the pharmacokinetics of tenofovir disoproxil fumarate (TDF), a potential concomitant medication for the treatment of HBV infection.
In vitro studies have shown that no clinically relevant interactions are predicted for efflux transporters (MDR1, BCRP, BSEP, MATE1, and MATE2K) and uptake transporters (OATP2B1, OAT1, OAT3, OCT1, and OCT2). In vitro bulevirtide inhibited the organic anion transporting polypeptides, OATP1B1 and OATP1B3, with IC50 values of 0.5 and 8.7 microM, respectively. Based on bulevirtide exposures, at the recommended 2 mg dose, no clinically relevant interaction is expected with OATP1B1 / OATP1B3 substrates.
In vitro studies have shown that bulevirtide (up to 5 microM) does not inhibit CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. No in vitro induction of CYP1A2, CYP2B6, or CYP3A4 by bulevirtide was observed.
Although anti-drug-antibodies (ADAs) for BLV appeared to have no effect on the efficacy or safety of BLV treatment, co-administration of BLV with PEG-IFN appeared to increase the incidence of ADAs compared to when BLV was administered alone.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Bulevirtide did not affect fertility or mating performance or early embryonic development in male and female rats at approximately 8.5 and 10 times higher exposures (AUC), respectively, than in humans given the recommended dose of Hepcludex.
(Category B1)
There are no adequate and well-controlled studies with Hepcludex in pregnant women. Hepcludex should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. In nonclinical reproductive toxicity studies, bulevirtide demonstrated no adverse effect on embryofetal development when administered to pregnant rats and rabbits at systemic exposures (AUC) 12- and 124-fold relative to exposure in humans at the recommended human dose.
It is not known whether bulevirtide is secreted in human milk. In nonclinical pre- and postnatal developmental rat studies, bulevirtide was not measured in the plasma of pups or in the milk of nursing animals.
A decision must be made whether to discontinue breast-feeding or to discontinue / abstain from treatment with bulevirtide, taking into account the benefit of breastfeeding for the child and the benefit of therapy for the woman.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects of Hepcludex on the ability to drive and use machines have been performed. Inform patients that dizziness has been reported during treatment with Hepcludex.
4.8 Adverse Effects (Undesirable Effects)
Experience from clinical studies.
Assessment of adverse reactions is based on pooled data from 64 patients with HDV who received 48 weeks of treatment with Hepcludex 2 mg in a Phase 2 study (MYR203) and a Phase 3 study (MYR301). The adverse reactions are listed in Table 1 by system organ class and frequency. Frequencies are defined as follows: very common (≥ 10%) or common (≥ 1% and < 10%).
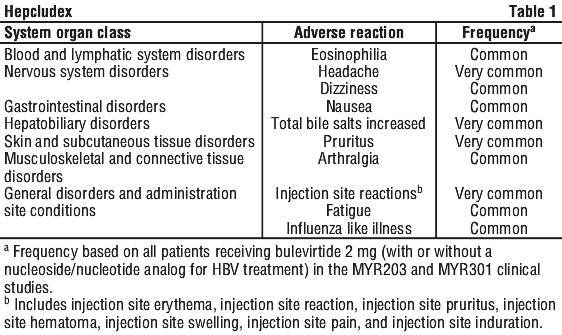 In a Phase 2 study (MYR202), 28 of 118 patients were randomised to receive once daily Hepcludex 2 mg (with TDF 300 mg for treatment of underlying HBV infection) for 24 weeks. The most common adverse reactions were headache, dizziness, and injection site reactions (grouped term includes injection site erythema and injection site rash).
In a Phase 2 study (MYR202), 28 of 118 patients were randomised to receive once daily Hepcludex 2 mg (with TDF 300 mg for treatment of underlying HBV infection) for 24 weeks. The most common adverse reactions were headache, dizziness, and injection site reactions (grouped term includes injection site erythema and injection site rash).
Laboratory changes. Eosinophil count increased.
Increases in eosinophil counts were commonly observed in patients receiving Hepcludex 2 mg; there were no associated clinical sequelae, hepatic adverse reactions, or significant liver-related laboratory abnormalities.
Total bile salts increased.
Asymptomatic bile salt elevations, associated with the mechanism of action of Hepcludex, were very commonly observed in clinical studies of Hepcludex 2 mg; the bile salt elevations resolved upon discontinuation of Hepcludex.
Due to renal excretion of bile salts, elevation of bile salts may be greater in patients with renal impairment.
Immunogenicity.
Hepcludex has the potential to induce antidrug antibodies (ADA), as detected in clinical studies using an enzyme-linked immunosorbent assay (ELISA). In Studies MYR203 and MYR301, a total of 64 patients who were treated with Hepcludex 2 mg monotherapy for 48 weeks were eligible for assessment of ADA prevalence; 18 of these patients (28.1%) were positive for ADA prevalence, of which 3 patients (4.7%) were positive for ADA at baseline. There is no evidence that the safety or effectiveness of Hepcludex were altered in these patients.
Postmarketing experience.
In addition to adverse reactions from clinical studies, the following adverse reactions were identified during post-approval use of Hepcludex. Because these reactions were reported voluntarily from a population of unknown size, estimates of frequency cannot be made.
Immune system disorders.
Hypersensitivity, including anaphylactic reaction.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There are no data on human overdose with bulevirtide. If overdose occurs, the patient must be monitored for evidence of toxicity and given standard supportive treatment as necessary.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Bulevirtide is a 47-amino acid, N-terminally myristoylated, HBV-L-protein derived, synthesized lipopeptide that blocks the entry of HBV and HDV into hepatocytes by binding to and inactivating the essential HBV and HDV entry receptor NTCP.
Antiviral activity.
In cell culture.
Bulevirtide (BLV) inhibited HDV infection in all the combinations of HBV and HDV genotypes tested in a primary human hepatocytes infectious system. The mean BLV EC50 values ranged from 0.26 to 0.64 nanoM across genotypes (GT) HDV-1 to HDV-8 and from 0.21 to 0.68 nanoM for HDV carrying envelopes across HBV GT A-H. Similarly, the mean BLV EC50 values against HDV-1 viruses pseudotyped with multiple strains of HBV GT-A to D were 0.57 nanoM (GTA), 0.59 nanoM (GTB), 0.43 nanoM (GTC), and 0.33 nanoM (GTD). For 137 clinical isolates, BLV had mean EC50 values of 0.40 nanoM, 0.45 nanoM, and 0.70 nanoM against HDV-1, HDV-5, and HDV-6, respectively. The mean EC50 values were 0.58 nanoM, 0.38 nanoM, and 0.45 nanoM against HDV clinical isolates carrying the envelopes from HBV GTA, GTD, and GTE, respectively.
Resistance.
In Study MYR301, resistance analysis was performed on 6 patients at Week 24 and 9 patients at Week 48 in BLV 2 mg group who experienced virologic breakthrough (2 consecutive increases in HDV RNA of ≥ 1 log10 IU/mL from nadir or 2 or more consecutive positive (target detected) HDV RNA values if previously HDV RNA was undetectable (target not detected) at 2 or more consecutive time points; 4 patients at Week 48) or HDV RNA decline < 1 log10 IU/mL (6 patients at Week 24 and 5 patients at Week 48). In Study MYR202, resistance analysis was performed on 5 patients in BLV 2 mg group who experienced virologic breakthrough (a single patient) or HDV RNA decline < 1 log10 IU/mL (4 patients) at Week 24. No amino acid substitutions tested at HBV BLV sequence positions or HDV HDAg associated with reduced susceptibility to Hepcludex were identified in these isolates from any of these patients at baseline, Week 24, and Week 48. All substitutions tested remained susceptible to bulevirtide in vitro. No resistance to Hepcludex was observed.
Clinical trials.
The efficacy of Hepcludex 2 mg once daily in the treatment of adults with chronic hepatitis D and compensated liver disease is based on data through Week 48 from one randomised, open-label Phase 3 study, Study MYR301 (N=150), and from data through Week 24 and Week 48 from two randomised open-label Phase 2 studies, Study MYR202 (N=118) and Study MYR203 (N=90), respectively. A total of 92 patients in Studies MYR301, MYR202, and MYR203 received Hepcludex 2 mg once daily. Patients were infected predominantly with GT HDV-1 consistent with the known dominant genotype of the virus.
Across Studies MYR301, MYR202, and MYR203, combined response was defined as undetectable HDV RNA or decrease in HDV RNA by ≥ 2 log10 IU/mL from baseline and ALT normalisation through Week 48 (MYR301 and MYR203) and through Week 24 (MYR202). Undetectable HDV RNA was defined as < lower limit of quantification [LLOQ] (target not detected) in Study MYR301; and < limit of detection [LOD], where LOD was 14 and 10 IU/mL in Studies MYR202 and MYR203, respectively.
MYR301.
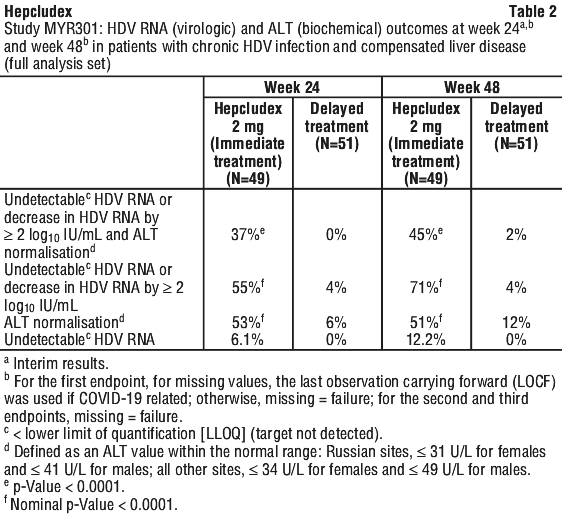
In Study MYR301, 100 of 150 patients with chronic HDV infection were randomised to receive immediate treatment with once daily Hepcludex 2 mg (N=49) or to have treatment delayed for 48 weeks (N=51). Randomization was stratified by the presence or absence of compensated cirrhosis. The proportion of participants achieving combined response at Week 48 was the defined primary endpoint.
Of the 49 patients in the immediate treatment group, mean age was 44 years; 61% were male, 84% were White, and 16% were Asian. Of the 51 patients in the delayed treatment group, mean age was 41 years; 51% were male, 78% were White, and 22% were Asian. All patients had infection with HDV genotype 1. Baseline characteristics were balanced among the immediate and delayed treatment groups. Of the patients in the immediate treatment group, at baseline, mean plasma HDV RNA was 5.1 log10 IU/mL, mean ALT was 108 U/L, 47% of patients had a history of cirrhosis, and 53% were interferon experienced. Patients were treated according to the standard care for their underlying HBV infection: the most common concomitant medications were TDF-containing or tenofovir alafenamide-containing products (49%) and entecavir (14%).
Table 2 presents the virologic and biochemical outcomes for immediate treatment with Hepcludex 2 mg once daily and delayed treatment at Week 24 and Week 48.
MYR202.
In Study MYR202, 56 of 118 patients with chronic HDV infection and ongoing viral replication who were interferon experienced, had a contraindication to interferon, or were cirrhotic, were randomised to receive Hepcludex 2 mg + TDF (N=28) or TDF alone (N=28) for 24 weeks. The proportion of participants achieving undetectable HDV RNA or a decrease by ≥ 2 log from baseline at Week 24 was defined as the primary endpoint.
Table 3 presents the virologic and biochemical outcomes at Week 24 in patients receiving Hepcludex 2 mg + TDF or TDF alone once daily.

MYR203.
In Study MYR203, 15 of 90 patients with chronic HDV infection were randomised to receive once daily Hepcludex 2 mg for 48 weeks. The proportion of patients with undetectable HDV RNA level at Week 72 (24 weeks after the end of the follow up period) was defined as the primary endpoint.
Table 4 presents the virologic and biochemical outcomes for immediate treatment with Hepcludex 2 mg once daily at Week 24 and Week 48.
 At 24 weeks following the end of treatment, 6.7% of patients had undetectable HDV RNA levels.
At 24 weeks following the end of treatment, 6.7% of patients had undetectable HDV RNA levels.
5.2 Pharmacokinetic Properties
The pharmacokinetic (PK) properties of bulevirtide were characterized after intravenous and subcutaneous administration. The exposure of bulevirtide increased in a more than proportional manner with increasing doses (dose range: 300 mcg to 20 mg intravenous; 800 mcg to 10 mg subcutaneous). Following 14 days of dosing, accumulation ratios for the recommended 2 mg dose for Cmax and AUC0-24h were approximately 2-fold. Based on clinical results and population PK analysis, no relationship could be identified between presence of ADA and bulevirtide PK. The steady state PK parameters of bulevirtide in Study MYR301 (based on population PK analysis) are provided in Table 5.

Absorption.
After subcutaneous injection, bulevirtide reached maximum plasma concentrations between 0.5 and 3 hours.
The absolute bioavailability of 2 mg bulevirtide after subcutaneous injection has not been estimated. Bioavailability following subcutaneous doses of 5 mg and 10 mg is estimated to be 48% and 57%, respectively. As bulevirtide demonstrates non-linear PK, extrapolation of bioavailability at other dose levels should be done with caution.
Distribution.
In vitro protein binding is high with > 99.9% of bulevirtide bound to plasma proteins. Following multiple dosing with bulevirtide 2 mg subcutaneous injection, the mean apparent volume of distribution was estimated to be 133 L in Study MYR203.
Biotransformation.
No biotransformation study was performed for bulevirtide. Bulevirtide is a linear peptide consisting of L-amino acids, and it is expected to be catabolized by peptidases to amino acids. No active metabolites are expected.
Excretion.
No bulevirtide excretion into urine was detected in healthy volunteers. Following multiple dosing with bulevirtide 2 mg subcutaneous injection, total mean apparent systemic clearance was estimated at 12.8 L/h in Study MYR203. After reaching peak concentrations, plasma levels declined with t1/2 of 3-7 hours.
Pharmacokinetics in special populations.
Age, sex, race, and body weight.
Based on population PK modeling, age, sex, race, or body weight did not have a clinically relevant impact on the systemic exposure of bulevirtide.
Hepatic impairment.
There is no clinically relevant impact of mild hepatic impairment (Child-Pugh A) on bulevirtide pharmacokinetics. Population PK modeling (n=154) characterized a 41.5% increase in AUCtau and 38.3% increase in Cmax in patients with mild hepatic impairment. The pharmacokinetics of bulevirtide have not been evaluated in patients with moderate and severe hepatic impairment (Child-Pugh B and C, respectively).
Renal impairment.
There is no impact of mild renal impairment (CrCl ≥ 60 and < 90 mL/min) on bulevirtide pharmacokinetics as confirmed by population PK modeling (n=60). The pharmacokinetics of bulevirtide have not been evaluated in patients with moderate and severe renal impairment (CrCl < 60 mL/min), or in patients with end-stage renal disease, including those on dialysis.
As bulevirtide is > 99.9% protein bound, dialysis is not expected to alter exposures of bulevirtide.
Geriatric patients.
The pharmacokinetics of bulevirtide have not been evaluated in the elderly (65 years of age and older).
5.3 Preclinical Safety Data
Genotoxicity.
Genotoxicity studies have not been conducted with bulevirtide due to the nature and mechanism of action of the product.
Carcinogenicity.
Carcinogenicity studies have not been conducted with bulevirtide.6 Pharmaceutical Particulars
6.1 List of Excipients
Mannitol, sodium carbonate, sodium bicarbonate, and may include hydrochloric acid and/or sodium hydroxide for pH adjustment.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
Information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
In order to protect from light, keep the vials in the outer carton.
Before reconstitution, the product should be stored in a refrigerator (2°C-8°C).
After reconstitution, chemical and physical in-use stability has been demonstrated for 2 hours at room temperature (up to 25°C). From a microbiological point of view, it is recommended that the product should be used immediately.
Product is for single use in one patient only. Discard any residue.
6.5 Nature and Contents of Container
Type 1 clear glass vial, an elastomeric closure, and an aluminum overseal with a flip-off cap.
Each carton contains 30 vials.
6.6 Special Precautions for Disposal
Any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.
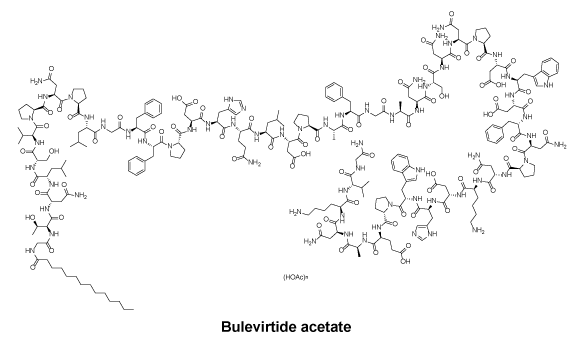
CAS number.
2012558-47-1.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
