What is in this leaflet
This leaflet answers some common questions about Hizentra®.
It does not contain all the available information. If you require further information about this medicine or your treatment, have any questions, or are not sure about something in this leaflet, consult your doctor or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the benefits that Hizentra® will have for you against the possible risks.
If you have any concerns about using this medicine, ask your doctor. Follow your doctor’s advice even if it is different from what this leaflet says.
Keep this leaflet with the medicine. You may need to read it again.
The information in this leaflet is subject to change.
Please check with your doctor whether there is any new information about this medicine that you should know since you were last treated.
What Hizentra® is used for
Your medicine is Hizentra®, a solution for subcutaneous infusion. Hizentra® contains human immunoglobulins. Immunoglobulins are also called antibodies and are a type of protein found in the blood. Immunoglobulins are produced by your body’s immune system to fight infections caused by bacteria and viruses. If you do not have enough antibodies you may not be able to fight off diseases.
Your doctor may give you Hizentra® either for:
- the replacement of antibodies because your antibody levels are low (referred to as immunodeficiency), or
- a condition where there is an imbalance in your immune system requiring treatment with immunoglobulins (referred to as immunomodulation).
Your doctor may have prescribed Hizentra® for another reason.
Ask your doctor if you have any questions about why it has been prescribed for you.
Before you are given Hizentra®
When you must not have it
Do not have Hizentra® if you have:
- a history of allergy to human immunoglobulin products (allergic reactions may include skin rash, face swelling, wheezing or breathing difficulties)
- previously been told you react to any of the ingredients in Hizentra® (the ingredients are presented in the last page on this leaflet under the heading Ingredients)
- been told you have antibodies to immunoglobulin A (IgA)
- too much proline in your blood (hyperprolinaemia).
If you are not sure whether you should be given this medicine, talk to your doctor.
Before you are given it
Tell your doctor if you:
- are pregnant or breast-feeding
- have had any vaccinations within the last two weeks
- are allergic to any medicine or food
- have IgA deficiency
- have a history of heart, or blood vessel disease, or blood clots, have thick blood, have been immobile for some time. Also tell the doctor what medicine you are using as some medicines such as those that contain the hormone estrogen (for example, birth control pills), may increase your risk of developing a blood clot.
- have any other medical conditions.
If you have not told your doctor about any of the above, tell them before you are given Hizentra®.
About blood products
When medicines are made from human blood or plasma, processes are used to prevent infections being passed from the blood/plasma donor to the person receiving the medicine. These processes include careful selection of the people who donate blood and plasma to make sure that those who might be carrying infections are excluded. In addition, each donation and pools of donations are tested for indicators of virus or virus infection(s).
Manufacturers of these medicines also include steps in the processing of blood or plasma that inactivate or remove viruses. Despite these processes, when medicines are prepared from human blood or plasma, the possibility of passing on an infection cannot be totally ruled out. Unknown or new viruses or other types of infection could also be passed on.
However, the measures taken in the manufacture of this medicine are considered effective for enveloped viruses such as human immunodeficiency virus (HIV), hepatitis B virus and hepatitis C virus, and for the non-enveloped viruses hepatitis A (HAV) and B19 virus (B19V).
There is reassuring clinical experience regarding the lack of HAV or B19V infections following treatment with immunoglobulin products. The antibodies which are in Hizentra® may also make an important contribution to limiting the possibility an infection could be passed on.
Please discuss the risks and benefits of this product with your doctor.
Taking other medicines
Tell your doctor if you are taking any other medicines, including medicines that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may affect the way other medicines work.
Vaccinations:
This product may affect how some vaccines work. Tell your doctor or healthcare professional that you are taking Hizentra® before you get a vaccine.
How to use Hizentra®
Treatment should be started and supervised by a doctor. Hizentra® is administered as an infusion subcutaneously (under the skin). If your doctor decides that you should receive Hizentra® at home, they will ensure you receive detailed instructions and training on how to use it.
If you do not understand the instructions ask your doctor or health professional.
How much is given
Your doctor will determine the dose(s) of Hizentra® that you will receive. Doses may be given at repeated intervals, from daily to once every two weeks. Your doctor may adjust the dose based on your response to the treatment.
Do not change the dose or dosing interval without consulting with your doctor. If you think you should receive Hizentra® more or less frequently, please speak to your doctor. If you think you have missed a dose, speak to your doctor as soon as possible.
How to prepare it
If your doctor considers that you should receive Hizentra® at home, the instructions below should be followed carefully.
Step 1: Clean surface
Clean a table or other flat surface.
Step 2: Assemble supplies
Gather the Hizentra® pre-filled syringe(s) or vial(s) and the following supplies (not provided with Hizentra®), as directed by your doctor: The Hizentra® vials/pre-filled syringes must be at room temperature before administration.
- Administration tubing
- Subcutaneous needle and required tubing
- Syringes
- Transfer device or drawing up needle(s)
- Gauze and tape, or transparent dressing (if required)
- Infusion pump (if required)
- Sharps container
- Alcohol (antiseptic) wipes
- Treatment diary.
Step 3: Wash hands
Thoroughly wash and dry your hands (Figure 1).

Step 4: Check vials or pre-filled syringes
If using pre-filled syringes, carefully peel back the transparent covering from the tray and inspect the protective cap. Peel back the outer layer of the wrap-around label to allow for viewing of Hizentra® through the fully transparent inner layer, but don’t remove the label completely (Figure 2).

If using vials, inspect the protective cap of the vials (Figure 3).

Hizentra® is a pale-yellow to light-brown clear solution. Check for particles or colour changes. Do not use the pre-filled syringe or vial if:
- the liquid looks cloudy, contains particles, or has changed colour.
- the protective cap of the pre-filled syringe or the vial is missing or defective.
- the expiry date on the label has passed.
Step 5: Preparation of Hizentra® for infusion
- If using Hizentra® pre-filled syringes, go to Step 5.1
- If using Hizentra® vials, go to Step 5.2
Step 5.1: Hizentra® pre-filled syringe(s)
The 5 mL and 10 mL pre-filled syringes are fully assembled and ready for use (Figure 4).
For the 20 mL and 50 mL pre-filled syringe, screw the plunger rod onto the pre-filled syringe stopper prior to use (Figure 5).


If you are using a syringe pump, Hizentra® pre-filled syringes can be placed directly in the syringe pump if the syringe size matches the pump requirements. Please follow the manufacturer’s instructions.
If the pre-filled syringe does not match the infusion pump requirements, transfer the contents of the pre-filled syringe to another syringe of a size specific for the infusion pump by following the directions below:
Use a syringe-to-syringe transfer device (tip-to-tip connector). (Figure 6)

Remove the protective cap from the pre-filled syringe. Attach the transfer device by twisting it onto the pre-filled syringe. Attach the empty syringe by screwing it onto the other side of the transfer device (Figure 7).

Push the plunger of the pre-filled syringe to transfer Hizentra® from the pre-filled syringe to the empty syringe.
- Repeat this step if multiple pre-filled syringes are necessary to achieve the prescribed dose. Remove the empty pre-filled syringe and attach another pre-filled syringe to the transfer device.
After the transfer is complete, remove the empty pre-filled syringe and the transfer device by unscrewing them from the syringe specific for your pump. Connect the filled syringe to the infusion tubing.
Go to Step 6.
Step 5.2: Transfer Hizentra® from vial(s) to syringe
Take the protective cap off the vial (Figure 8).

Clean the vial stopper with an alcohol wipe (Figure 9). Allow to dry.

Attach a needle or transfer device to the syringe tip, using a clean (non-touch) technique. If using a transfer device, follow the instructions provided by the device manufacturer. If using a needle and a syringe to transfer Hizentra®, follow the instructions below.
- Attach a sterile transfer needle to a sterile syringe (Figure 10).

- Pull out the plunger of the syringe to fill the syringe with air. Make sure the amount of air is the same as the amount of Hizentra® you will transfer from the vial.
- Put the Hizentra® vial on a flat surface. Keeping the vial upright, insert the transfer needle into the centre of the rubber stopper.
- Check that the tip of the needle is not in the liquid. Then, push the plunger of the syringe down. This will inject the air from the syringe into the airspace of the vial.
- Leaving the needle in the stopper while keeping pressure on the syringe plunger, carefully turn the vial upside down (Figure 11).

- Gently withdraw the needle so the tip sits in the Hizentra® fluid and release the pressure on the plunger. The fluid will automatically fill the syringe. Once the automatic filling is complete slowly pull back on the plunger to fill the syringe with any remaining Hizentra®.
- Take the filled syringe and needle out of the stopper. Take off the needle and throw it away in the sharps container.
- When using multiple vials to achieve the desired dose, repeat this step.
Step 6: Prepare the Subcutaneous needles and related tubing
Attach the subcutaneous needle to infusion tubing (if required).
Prime (fill) the infusion tubing. To prime the tubing, connect the syringe filled with Hizentra® to the infusion tubing and gently push on the syringe plunger to fill the tubing with Hizentra® (Figure 12).

Stop priming before Hizentra® fluid reaches the needle.
Step 7: Prepare infusion site(s)
Select an area on your abdomen, thigh, upper arm, or side of upper leg/hip area for the infusion (Figure 13). Never infuse into areas where the skin is tender, bruised, red, or hard. Avoid infusing into scars or stretch marks. The number of infusion sites depends on the volume of the total dose. Your healthcare professional can advise you about this.

If you are using more than one infusion site, be sure each site is at least 5 cm apart.
Clean the skin at each site with an alcohol wipe (Figure 14). Let the skin dry.
Step 8: Insert needle(s)
Using two fingers, pinch the skin around the infusion site. Insert the needle into the skin (Figure 15). Your healthcare professional will advise you on the positioning of the needle as it is dependent on the type of needle used.

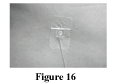
Secure the needle in place as directed by your healthcare professional (Figure 16). This will keep the needle from coming out.
Step 9: Start infusion
MAKE SURE YOU ARE NOT INFUSING HIZENTRA® INTO A BLOOD VESSEL. To test for this, pull the plunger back gently. If you see any blood flowing back into the tubing, remove the needle. Discard the tubing and needle and restart from Step 6.
If using an infusion pump, prepare the pump following the manufacturer’s instructions.
Follow the instructions that you have been given by your healthcare professional to start the infusion. Infuse at the rate you have been instructed (Figure 17).

Step 10: Record treatment
Complete your treatment diary as instructed by your healthcare professional.
Step 11: Clean up
Remove the needle set and cover the infusion site with a protective dressing as directed by your healthcare professional.
Discard the empty Hizentra® pre-filled syringe(s) or vial(s), needles, infusion tubing, and syringes as directed by your healthcare professional (Figure 18).

General rubbish such as packaging can be discarded in the general waste.
If using an infusion pump, clean and store it, following the manufacturer’s instructions.
If too much is given (overdose)
The effects of an overdose of Hizentra® are not known.
Please tell your doctor if you accidently use more than instructed.
While you are having Hizentra®
This medicine is not expected to affect your ability to drive a car or operate machinery.
Things you must do
Tell your doctor if you are planning to have a vaccination. Hizentra® may impair the effect of some virus vaccines such as measles, mumps, rubella and chicken pox for a period of at least six weeks, and up to three months. After receiving this medicine, a period of three months should be allowed before vaccination with some virus vaccines. In the case of measles vaccine, this effect may last for up to one year, so if you are going to receive a measles vaccine you should have your measles antibody status checked.
Tell your doctor if you are about to have any blood tests. Hizentra® may interfere with the results of some tests, resulting in misleading results for some things.
Inform other doctors, dentists, and pharmacists who treat you that you have been given this medicine. It is important for them to know if they are starting you on any other new medicines.
Tell your doctor if you are planning to travel overseas. It is important to obtain a written statement from your doctor explaining the reasons why you need to have this medicine and infusing devices with you, otherwise you may not be allowed to bring it into the country of travel. Please ensure you have multiple copies of the letter if travelling to more than one country.
Side effects
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you experience some of the side effects.
Do not be alarmed by the following lists of possible side effects. You may not experience any of them. If you have any questions, ask your doctor.
Tell your doctor immediately or go to the Accident and Emergency Department at your nearest hospital if you notice any of the following symptoms:
- reduced urination
- severe headache
- neck stiffness
- inability to stand bright light
- painful eye movements
- pain/tenderness, swelling/discolouration of an arm or leg
- tingling, numbness or weakness on one side of the body
- shortness of breath
- chest pain
- fever
- allergic or anaphylactic reaction, symptoms of which may include:
- swelling of the lips, tongue or eyes
- loss of consciousness
- hives
- difficulty in breathing.
Tell your doctor if you notice any of the following and they worry you.
This list includes the more common side effects of Hizentra®. They are usually mild and often reduce over time.
- swelling, pain, redness or itching where the infusion was given
- headache/migraine
- nausea or vomiting
- pain (including pain in the chest, back, joints, arms, legs)
- muscle pain
- fatigue
- diarrhoea
- abdominal pain
- fever or chills
- feeling faint, dizzy or light headed (fall in blood pressure)
- infusion site ulcer.
Other side effects not listed above may also occur in some patients.
Tell your doctor if you notice any other effects.
After having Hizentra®
Storing Hizentra®
Store Hizentra® in a cool dry place where the temperature stays below 25°C. Do not freeze.
Keep the vial or pre-filled syringe in the outer carton in order to protect from light.
Do not use after the expiry date.
Keep it out of the sight and reach of children.
Product description
What it looks like
Hizentra® is a clear and colourless or pale-yellow to light-brown solution. It is packaged in single-use clear glass vials or single-use pre-filled syringes.
Ingredients
Each vial or pre-filled syringe of Hizentra® contains a sterile solution comprising 20% plasma proteins of which at least 98% are immunoglobulins. This medicine contains proline, polysorbate 80 and water. Hizentra® does not contain any preservatives, so any unused portion should be discarded immediately. Hizentra® is packaged using latex free materials.
Distributor
CSL (Behring) Australia Pty Ltd
ABN 48 160 734 761
189–209 Camp Road
Broadmeadows VIC 3047
Date of most recent amendment
February 2025
Australian Register Numbers
Vials:
1 g in a 5 mL solution: AUST R 207386
2 g in a 10 mL solution: AUST R 207385
4 g in a 20 mL solution: AUST R 207383
10 g in a 50 mL solution: AUST R 207384
Pre-filled syringes:
1 g in a 5 mL solution: AUST R 285344
2 g in a 10 mL solution: AUST R 285345
4 g in a 20 mL solution: AUST R 329342
10 g in a 50 mL solution: AUST R 457854
® Registered trademark of CSL Limited Group of Companies
Published by MIMS May 2025
 It is recommended to use needles gauge 24 or larger (i.e. lower gauge number). Using smaller needles (i.e. higher gauge number) may make it more difficult to manually push Hizentra.
It is recommended to use needles gauge 24 or larger (i.e. lower gauge number). Using smaller needles (i.e. higher gauge number) may make it more difficult to manually push Hizentra.
 Simulations with empirical population-pharmacokinetic models suggested that a comparable IgG exposure (AUC0-14 days, Cmin 14 days) is achieved when Hizentra is administered subcutaneously every two weeks using double the weekly dose during maintenance therapy. These simulations further suggested that comparable serum IgG trough levels are achieved when the weekly maintenance dose of Hizentra is administered in proportional amounts more frequently than once a week (e.g. 2 times per week, 3 times per week, 5 times per week or daily).
Simulations with empirical population-pharmacokinetic models suggested that a comparable IgG exposure (AUC0-14 days, Cmin 14 days) is achieved when Hizentra is administered subcutaneously every two weeks using double the weekly dose during maintenance therapy. These simulations further suggested that comparable serum IgG trough levels are achieved when the weekly maintenance dose of Hizentra is administered in proportional amounts more frequently than once a week (e.g. 2 times per week, 3 times per week, 5 times per week or daily).