What is in this leaflet
This leaflet answers some common questions about IBRANCE.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking IBRANCE against the benefits it is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What IBRANCE is used for
What IBRANCE does
IBRANCE is used to treat patients with hormone receptor-positive (HR-positive), human epidermal growth factor receptor 2 negative (HER2-negative) advanced breast cancer. It is given together with an aromatase inhibitor or fulvestrant, which are used as hormonal anticancer therapies.
IBRANCE belongs to a group of medicines called cyclin-dependent kinase inhibitors.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
IBRANCE is only available with a doctor's prescription.
It is not addictive.
Use in Children
The safety and efficacy of IBRANCE in children have not been established.
Before you take IBRANCE
When you must not take it
Do not take IBRANCE if you have an allergy to:
- any medicine containing palbociclib
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have a fever, chills or any other signs or symptoms of infection.
Tell your doctor if you have or have had any of the following medical conditions:
- abnormal blood test results
- kidney problems
- liver problems
- lactose intolerance.
You should have a blood test before starting treatment with IBRANCE.
Tell your doctor if you are pregnant or plan to become pregnant. IBRANCE may affect the developing baby and should not be taken during pregnancy. It is recommended you use contraception whilst you take IBRANCE. Your doctor will discuss the risks with you.
IBRANCE may decrease fertility in men. Men may consider preserving sperm before taking IBRANCE.
Tell your doctor if you are breast-feeding. You should not breast feed if you are being treated with IBRANCE.
If you have not told your doctor about any of the above, tell him/her before you start taking IBRANCE.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including:
- all prescription medicines
- all medicines, vitamins, herbal supplements or natural therapies you buy without a prescription from a pharmacy, supermarket, naturopath or health food shop.
Some medicines may be affected by IBRANCE or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines. Your doctor will advise you.
Tell your doctor or pharmacist if you are taking any of the following:
- medicines used to treat fungal infections such as posaconazole, voriconazole, ketoconazole, miconazole or itraconazole
- antibiotics used to treat bacterial infections such as erythromycin or clarithromycin
- medicines used to treat HIV infections/AIDS such as atazanavir, indinavir, efavirenz, ritonavir, lopinavir, fosamprenavir, nevirapine, etravirine or saquinavir
- medicines used to treat hepatitis C such as elbasvir/grazoprevir
- medicines used to treat tuberculosis (TB) such as rifampin or rifabutin,
- medicines used to treat certain heart conditions or high blood pressure such as bosentan or diltiazem
- medicines used to treat epilepsy, seizures or fits such as phenytoin, carbamazepine, felbamate, primidone or phenobarbital
- St John's wort (Hypericum perforatum), a herbal medicine used to treat depression and other conditions
- nefazodone, used to treat depression
- modafinil, used to treat sleep disorders
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take IBRANCE
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you which strength of IBRANCE you need to take. This may depend on your age, your condition and whether or not you are taking any other medicines.
For advanced breast cancer the recommended dose of IBRANCE is one 125 mg capsule or tablet taken once daily for 21 days followed by 7 days without taking IBRANCE.
Your doctor may change your dose during treatment.
Taking IBRANCE for 21 continuous days followed by 7 days without taking IBRANCE (total of 28 days) is counted as one treatment cycle.
The 7 day break when you are not taking IBRANCE helps your body recover. It reduces your chance of getting side effects and could stop you getting an infection.
How to take it
Swallow the IBRANCE capsule or tablet whole with a glass of water. Do not chew, crush, open the capsules or split the tablets prior to swallowing.
When to take it
You should take IBRANCE capsules with food and at the same time each day. Taking it with food and at the same time each day will have the best effect. It will also help you remember when to take it.
You should take IBRANCE tablets at the same time each day and can take it with or without food.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it or vomit after taking a dose
Take your next dose at your regular time.
Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor, or Poisons Information Centre (telephone Australia 13 11 26 or New Zealand 0800 POISON or 0800 764 766) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much IBRANCE.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using IBRANCE
Things you must do
Make sure you follow your doctor's instructions and keep all appointments.
You should have a blood test before each treatment cycle. The blood test is done to make sure your blood cells have recovered from the last treatment cycle and to check for side effects.
Tell your doctor immediately if you experience signs or symptoms of an infection such as fever and chills. IBRANCE may reduce the number of your white blood cells and weaken your immune system. You may be at greater risk of getting an infection while you are taking IBRANCE.
Use contraception (birth control) to prevent pregnancy while you are being treated with IBRANCE. Women who could become pregnant or men who could father a child must use a reliable method of contraception during treatment with IBRANCE. Women should continue using contraception for at least a month after taking their last dose of IBRANCE and males should continue using contraception for 14 weeks after the last dose of IBRANCE.
Tell your doctor immediately if you become pregnant while taking IBRANCE.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking IBRANCE.
Things you must not do
Do not eat grapefruit or drink grapefruit juice while you are being treated with IBRANCE. Grapefruit and grapefruit juice may change the amount of IBRANCE in your body.
Do not take IBRANCE to treat any other complaints unless your doctor tells you to.
Do not give IBRANCE to anyone else, even if they have the same condition as you.
Do not stop taking IBRANCE or lower the dosage without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how IBRANCE affects you. This medicine may cause fatigue and blurred vision in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Side effects
Like all medicines, IBRANCE can cause side effects.
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking IBRANCE.
Do not be alarmed by the list of side effects. You may not experience any of them.
Tell your doctor if...
Tell your doctor or pharmacist if you notice any of the following:
- tiredness
- nausea (feeling sick) or vomiting
- diarrhoea
- sore mouth, lips or tongue
- hair loss
- loss of appetite
- nose bleed
- skin rash
- change in sense of taste
- blurred vision, increased tearing or dry eyes
Go to hospital if...
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital, if you notice any of the following:
- infection, fever, chills
- mouth ulcers, sore throat, sore gums
- lack of energy, pale skin, shortness of breath
- weakness, dizziness, rapid heart rate
- bleeding or bruising more easily than usual
The above side effects may be serious. You may need urgent medical attention or hospitalisation.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some people.
Some side effects can only be found when your doctor does tests to check your progress. These tests could show a reduction in white blood cells, red blood cells or platelets. These tests could also show a change in liver enzymes.
After using IBRANCE
Storage
Keep your IBRANCE in the original pack until it is time to take it. If you take the capsules or tablets out of the pack they may not keep well.
Keep your IBRANCE pack in a cool dry place where the temperature stays below 30°C.
Do not store IBRANCE or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
IBRANCE 75 mg:
Capsules: light orange cap and body printed with "Pfizer" on the cap and "PBC 75" on the body in white ink.
Tablets: round, light purple, film-coated tablet debossed with “Pfizer” on one side and “PBC 75” on the other side.
IBRANCE 100 mg:
Capsules: caramel cap and light orange body printed with "Pfizer" on the cap and "PBC 100" on the body in white ink.
Tablets: oval, green, film-coated tablet debossed with “Pfizer” on one side and “PBC 100” on the other side.
IBRANCE 125 mg:
Capsules: caramel cap and body printed with "Pfizer" on the cap and "PBC 125" on the body in white ink.
Tablets: oval, light purple, film-coated tablet debossed with “Pfizer” on one side and “PBC 125” on the other side.
Each capsule blister pack or bottle contains 21 capsules.
Each tablet blister pack contains 21 film-coated tablets.
Ingredients
IBRANCE capsules and tablets contain 75 mg, 100 mg or 125 mg of palbociclib as the active ingredient.
The capsules also contain:
- microcrystalline cellulose
- lactose monohydrate
- sodium starch glycollate
- silicon dioxide
- magnesium stearate
- gelatin
- iron oxide red (E172)
- iron oxide yellow (E172)
- titanium dioxide (E171)
The tablets have a tablet core containing:
- microcrystalline cellulose
- silicon dioxide
- crospovidone
- magnesium stearate
- succinic acid
The tablets also have a film coating containing:
- hypromellose
- titanium dioxide (E171)
- triacetin
- indigo carmine aluminium lake (E132)
- iron oxide red (E172)
- iron oxide yellow (E171)
IBRANCE does not contain sucrose, gluten, tartrazine or any other azo dyes.
Supplier
IBRANCE is supplied in Australia by:
Pfizer Australia Pty Ltd
Sydney, NSW
Toll Free number: 1800 675 229
Australian registration numbers
IBRANCE 75 mg:
AUST R 274622 (capsule blister)
AUST R 274624 (capsule bottle)
AUST R 319780 (tablet blister)
IBRANCE 100 mg:
AUST R 274623 (capsule blister)
AUST R 274621 (capsule bottle)
AUST R 319782 (tablet blister)
IBRANCE 125 mg:
AUST R 274619 (capsule blister)
AUST R 274620 (capsule bottle)
AUST R 319784 (tablet blister)
Date of preparation
This leaflet was prepared in June 2020.
® = Registered Trademark
© Pfizer Australia Pty Ltd
Published by MIMS August 2020


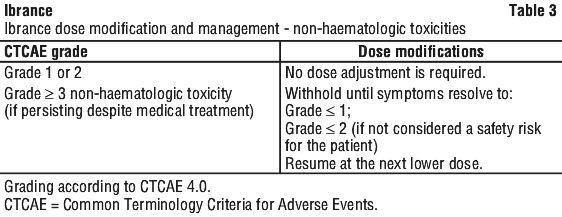 Permanently discontinue Ibrance in patients with severe interstitial lung disease (ILD)/pneumonitis (see Section 4.4 Special Warnings and Precautions for Use, Interstitial lung disease (ILD)/pneumonitis).
Permanently discontinue Ibrance in patients with severe interstitial lung disease (ILD)/pneumonitis (see Section 4.4 Special Warnings and Precautions for Use, Interstitial lung disease (ILD)/pneumonitis).
 The most common adverse reactions (≥ 20%) of any grade reported in patients in the Ibrance plus letrozole arm by descending frequency were neutropenia, infections, leukopenia, nausea, fatigue, alopecia, stomatitis, anaemia, diarrhoea and thrombocytopenia.
The most common adverse reactions (≥ 20%) of any grade reported in patients in the Ibrance plus letrozole arm by descending frequency were neutropenia, infections, leukopenia, nausea, fatigue, alopecia, stomatitis, anaemia, diarrhoea and thrombocytopenia.
 The most common adverse drug reactions of any grade reported in > 20% of patients receiving palbociclib in combination with fulvestrant were neutropenia, leukopenia, infections, fatigue, nausea, anaemia, stomatitis, diarrhoea, thrombocytopenia and vomiting.
The most common adverse drug reactions of any grade reported in > 20% of patients receiving palbociclib in combination with fulvestrant were neutropenia, leukopenia, infections, fatigue, nausea, anaemia, stomatitis, diarrhoea, thrombocytopenia and vomiting. The primary endpoint of the study was PFS evaluated according to RECIST version 1.1 as assessed by investigator. Secondary efficacy endpoints included objective response (OR), duration of response (DOR), clinical benefit response (CBR), overall survival (OS), safety, EQ-5D scores and health related quality of life (QoL) assessed using the Functional Assessment of Cancer Therapy Breast (FACT-B) questionnaire.
The primary endpoint of the study was PFS evaluated according to RECIST version 1.1 as assessed by investigator. Secondary efficacy endpoints included objective response (OR), duration of response (DOR), clinical benefit response (CBR), overall survival (OS), safety, EQ-5D scores and health related quality of life (QoL) assessed using the Functional Assessment of Cancer Therapy Breast (FACT-B) questionnaire. The Kaplan-Meier curves for PFS based on the updated cutoff date of 31 May 2017 are displayed in Figure 1.
The Kaplan-Meier curves for PFS based on the updated cutoff date of 31 May 2017 are displayed in Figure 1. Prespecified subgroup analyses indicated that the treatment effect (by median PFS) was consistent in all subgroups defined by stratification factors and baseline characteristics, including presence/absence of visceral metastases at baseline and presence of bone-only disease at baseline. An exploratory analysis showed that median PFS was longer in the palbociclib plus letrozole arm for 512 patients whose tumour tested positive for Rb protein expression by immunohistochemistry (IHC) (HR 0.543 [95% CI: 0.433, 0.681], mPFS 27.4 months versus 13.7 months). For the 51 patients whose tumours tested negative for Rb protein expression by IHC, the difference between treatment arms was not statistically significant.
Prespecified subgroup analyses indicated that the treatment effect (by median PFS) was consistent in all subgroups defined by stratification factors and baseline characteristics, including presence/absence of visceral metastases at baseline and presence of bone-only disease at baseline. An exploratory analysis showed that median PFS was longer in the palbociclib plus letrozole arm for 512 patients whose tumour tested positive for Rb protein expression by immunohistochemistry (IHC) (HR 0.543 [95% CI: 0.433, 0.681], mPFS 27.4 months versus 13.7 months). For the 51 patients whose tumours tested negative for Rb protein expression by IHC, the difference between treatment arms was not statistically significant. An analysis of time to deterioration composite endpoint (TTD) in Functional Assessment of Cancer Therapy Breast (FACT-B), defined as the time between baseline and first occurrence of decrease of ≥ 7 points in FACT-B scores, was carried out based on survival analysis methods using a Cox proportional hazards model and log rank test. No statistically significant difference was observed in TTD in FACT-B total scores between the palbociclib plus letrozole arm and the placebo plus letrozole arm (HR of 1.042 [95% CI: 0.838, 1.295]; 1-sided p-value = 0.663).
An analysis of time to deterioration composite endpoint (TTD) in Functional Assessment of Cancer Therapy Breast (FACT-B), defined as the time between baseline and first occurrence of decrease of ≥ 7 points in FACT-B scores, was carried out based on survival analysis methods using a Cox proportional hazards model and log rank test. No statistically significant difference was observed in TTD in FACT-B total scores between the palbociclib plus letrozole arm and the placebo plus letrozole arm (HR of 1.042 [95% CI: 0.838, 1.295]; 1-sided p-value = 0.663).

 The primary endpoint of the study was investigator assessed PFS evaluated according to RECIST version 1.1. Secondary PFS analyses were based on a random sample Blinded Independent Central Radiology Review (BICR) of 40.5% of the ITT population. Secondary endpoints included OR, DOR, CBR, OS, safety, change in QoL and TTD. Patient reported outcomes including Global QoL and pain were measured using the European Organisation for Research and Treatment of Cancer (EORTC) quality of life questionnaire (QLQ-C30) and the Breast Cancer Module (BR23) questionnaire.
The primary endpoint of the study was investigator assessed PFS evaluated according to RECIST version 1.1. Secondary PFS analyses were based on a random sample Blinded Independent Central Radiology Review (BICR) of 40.5% of the ITT population. Secondary endpoints included OR, DOR, CBR, OS, safety, change in QoL and TTD. Patient reported outcomes including Global QoL and pain were measured using the European Organisation for Research and Treatment of Cancer (EORTC) quality of life questionnaire (QLQ-C30) and the Breast Cancer Module (BR23) questionnaire.
 Prolongation of PFS in the palbociclib plus fulvestrant arm was also demonstrated in most individual patient subgroups supporting internal consistency of PFS benefit findings within the study and was supported by the secondary PFS random sample BICR audit. See Figure 4.
Prolongation of PFS in the palbociclib plus fulvestrant arm was also demonstrated in most individual patient subgroups supporting internal consistency of PFS benefit findings within the study and was supported by the secondary PFS random sample BICR audit. See Figure 4. As per inclusion criteria, pre/perimenopausal women were enrolled in the study if already treated with an LHRH agonist from at least 4 weeks. If they were not treated with goserelin prior to study entry, they were switched to goserelin when they started the study treatment.
As per inclusion criteria, pre/perimenopausal women were enrolled in the study if already treated with an LHRH agonist from at least 4 weeks. If they were not treated with goserelin prior to study entry, they were switched to goserelin when they started the study treatment.
 A positive treatment effect of palbociclib plus fulvestrant versus placebo plus fulvestrant on OS was observed in the majority of the prespecified subgroups. Due to the low event number and smaller sample size in some of the prespecified subgroups, the magnitude of estimated effect of palbociclib added to fulvestrant could not always be determined. The OS results from patients subgroups defined by stratification factors at randomisation are reported in Table 15.
A positive treatment effect of palbociclib plus fulvestrant versus placebo plus fulvestrant on OS was observed in the majority of the prespecified subgroups. Due to the low event number and smaller sample size in some of the prespecified subgroups, the magnitude of estimated effect of palbociclib added to fulvestrant could not always be determined. The OS results from patients subgroups defined by stratification factors at randomisation are reported in Table 15. The estimated survival probabilities for palbociclib plus fulvestrant versus placebo plus fulvestrant were respectively: 65.3% (95% CI: 59.9, 70.2) vs. 57.3% (95% CI: 49.2, 64.6) at 2 years and 49.6% (95% CI: 44.0, 54.9) vs. 40.8% (95% CI: 32.9, 48.5) at 3 years.
The estimated survival probabilities for palbociclib plus fulvestrant versus placebo plus fulvestrant were respectively: 65.3% (95% CI: 59.9, 70.2) vs. 57.3% (95% CI: 49.2, 64.6) at 2 years and 49.6% (95% CI: 44.0, 54.9) vs. 40.8% (95% CI: 32.9, 48.5) at 3 years. Palbociclib is a yellow to orange powder with a pKa of 7.4 (secondary piperazine nitrogen) and 3.9 (pyridine nitrogen). The solubility of palbociclib in aqueous media decreases over the range pH 4.3 to pH 9.0 from greater than 0.7 mg/mL to less than 0.002 mg/mL. At or below pH 4, palbociclib behaves like a high solubility compound. Above pH 4, the solubility of the drug substance reduces significantly. The partition coefficient (1-octanol/water) at pH 7.4 is 0.99.
Palbociclib is a yellow to orange powder with a pKa of 7.4 (secondary piperazine nitrogen) and 3.9 (pyridine nitrogen). The solubility of palbociclib in aqueous media decreases over the range pH 4.3 to pH 9.0 from greater than 0.7 mg/mL to less than 0.002 mg/mL. At or below pH 4, palbociclib behaves like a high solubility compound. Above pH 4, the solubility of the drug substance reduces significantly. The partition coefficient (1-octanol/water) at pH 7.4 is 0.99.