1 Name of Medicine
Eflornithine.
2 Qualitative and Quantitative Composition
Each tablet contains 192 mg eflornithine (as hydrochloride).
Eflornithine may also be known as difluoromethylornithine (DFMO).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Tablet.
White to off white, round, biconvex tablets with a dimension of approximately 11 mm, debossed with "EFL" on one side and "192" debossed on the other side.
4.1 Therapeutic Indications
Ifinwil is indicated for the treatment of adults and paediatric patients with high-risk neuroblastoma (HRNB) who have responded to prior multiagent, multimodality therapy.
4.2 Dose and Method of Administration
Treatment should be initiated and overseen by a physician who is experienced in the treatment of oncology. Prior to initiating treatment with Ifinwil, perform a full blood count, liver function tests and baseline hearing assessments (see Section 4.4 Special Warnings and Precautions for Use).
Dose.
The recommended dose of Ifinwil depends on body surface area (BSA) as detailed in Table 1. Recalculate the appropriate dose every 3 months (as BSA changes) during treatment.
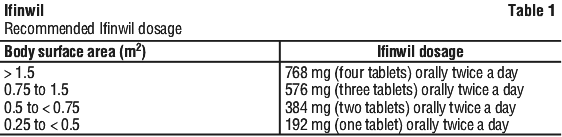 Continue treatment for 2 years, or until recurrence of disease or unacceptable toxicity occurs.
Continue treatment for 2 years, or until recurrence of disease or unacceptable toxicity occurs.
Dosage modifications for adverse reactions.
For patients with adverse reactions who require dose reduction, the recommended dose reduction levels are provided in Table 2.
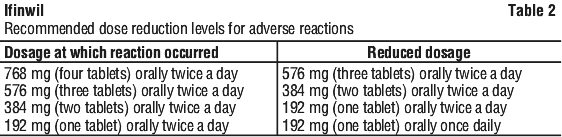 If subsequent adverse reactions occur, continue dose reduction until reaching the minimum dose of one 192 mg tablet once per day. Permanently discontinue Ifinwil if the patient is unable to tolerate the minimum dose of 192 mg once daily.
If subsequent adverse reactions occur, continue dose reduction until reaching the minimum dose of one 192 mg tablet once per day. Permanently discontinue Ifinwil if the patient is unable to tolerate the minimum dose of 192 mg once daily.
Recommended dosage modifications of Ifinwil for the management of adverse reactions are provided in Table 3.

Dosing in special populations.
Paediatric population.
The safety and efficacy of Ifinwil in children under 1 year has not been established. No data are available.
Renal impairment.
Renal impairment affects the pharmacokinetics of Ifinwil (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties). For patients with severe renal impairment (eGFR ≤ 29 mL/min/1.73 m2, reduce the dose by 50% as described in Table 4.

Hepatic impairment.
Based on population pharmacokinetic analysis, no dose adjustment is required for patients with mild hepatic impairment. Ifinwil has not been studied in patients with moderate or severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Method of administration.
Ifinwil tablets are taken orally, with or without food, and can be swallowed whole, chewed or crushed.
Crushed administration.
For crushed administration, mix crushed Ifinwil tablets with a small amount (approximately 2 tablespoons or 30 mL) of soft food or liquid. Visually confirm the entire contents have been consumed. If any crushed tablet particles remain in the container, mix with an additional small amount (e.g. no more than 30 mL) of soft food or liquid and ensure all remaining particles of crushed tablet are consumed. Discard crushed tablet preparation after one hour.
Missed dose.
A missed dose should be taken as soon as possible. However, if the next dose is due to be taken within the next 7 hours, the missed dose should be skipped.
If vomiting occurs after taking a dose, an additional dose should not be taken. Continue with the next scheduled dose.4.3 Contraindications
Hypersensitivity to eflornithine or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Myelosuppression.
Ifinwil can cause myelosuppression. In a pooled safety population (see Section 4.8 Adverse Effects (Undesirable Effects)), Grade 3 or 4 neutropenia occurred in 4.2% of patients. Febrile neutropenia occurred in 0.6% of patients. Bone marrow failure occurred in 1 patient. Grade 3 or 4 thrombocytopenia occurred in 1.4% of patients. Grade 3 anaemia occurred in 3.3% of patients.
Monitor blood counts including neutrophil count, platelet count, and haemoglobin level prior to administration of Ifinwil and periodically during treatment. Withhold, reduce the dose, or permanently discontinue Ifinwil based on severity (see Section 4.2 Dose and Method of Administration, Table 3).
Hepatotoxicity.
Ifinwil can cause hepatotoxicity. In the pooled safety population (see Section 4.8 Adverse Effects (Undesirable Effects)), Grade 3 or 4 events of increased alanine aminotransferase (ALT) occurred in 11% of patients. Grade 3 or 4 events of increased aspartate aminotransferase (AST) occurred in 6% of patients. Grade 3 or 4 events of increased bilirubin occurred in 0.3% of patients. Increased ALT/AST leading to dose interruption or reduction occurred in 2.5% of patients. Ifinwil was discontinued due to increased ALT/AST in 0.6% of patients.
Perform liver function tests (ALT, AST, and total bilirubin) prior to the start of Ifinwil, every month for the first six months of treatment, then once every 3 months or as clinically indicated, with more frequent testing in patients who develop transaminase or bilirubin elevations. Withhold and reduce the dose or permanently discontinue Ifinwil based on severity (see Section 4.2 Dose and Method of Administration, Table 3).
Hearing loss/hypoacusis.
Ifinwil can cause hearing loss. In the pooled safety population (see Section 4.8 Adverse Effects (Undesirable Effects)), 81% of patients had an abnormal audiogram at baseline. New or worsening hearing loss occurred in 13% of patients who received Ifinwil; hearing loss worsened from baseline to Grade 3 or 4 in 12% of patients. Tinnitus occurred in 1 patient. Hearing loss leading to dose interruption or reduction occurred in 4% of patients. New or worsening hearing loss requiring new use of hearing aids occurred in 7% of patients. Ifinwil was discontinued due to hearing loss in 1.4% of patients. Among all patients with new or worsening hearing loss during Ifinwil treatment, the hearing loss resolved to baseline in 9% of patients. Among 18 patients who experienced new or worsening hearing loss and had dose modifications, 67% (N=12) improved or resolved to baseline.
Perform audiogram prior to initiation of therapy and at 6-month intervals, or as clinically indicated, to monitor for potential hearing loss. Withhold and reduce the dose or permanently discontinue Ifinwil based on severity (see Section 4.2 Dose and Method of Administration, Table 3).
Use in renal impairment.
Renal impairment affects the pharmacokinetics of Ifinwil (see Section 5.2 Pharmacokinetic Properties). Use Ifinwil with caution in patients with moderate renal impairment (estimated glomerular filtration rate [GFR] 30-59 mL/min/1.73 m2). Monitor closely for adverse reactions including hepatotoxicity, myelosuppression, and hearing loss, and adjust dosage if appropriate (see Section 4.2 Dose and Method of Administration, Table 3).
For patients with severe renal impairment (eGFR ≤ 29 mL/min/1.73 m2), reduce the dose by 50% as described in Section 4.2 Dose and Method of Administration, Table 4.
Use in the elderly.
No data available.
Paediatric use.
The use of Ifinwil to treat paediatric patients with HRNB who have responded to prior multiagent, multimodality therapy is supported by evidence from adequate and well-controlled studies in patients aged 1-17 years, with a median age of 4 years (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.2 Pharmacokinetic Properties; Section 5.1 Pharmacodynamic Properties, Clinical efficacy). The safety and effectiveness of Ifinwil have not been established in paediatric patients for other indications.
Effects on laboratory tests.
See Section 4.8 Adverse Effects (Undesirable Effects), Table 6.4.5 Interactions with Other Medicines and Other Forms of Interactions
No in-vivo drug interaction studies have been performed with other medicinal products.
In vitro studies showed eflornithine is not a substrate or inhibitor of cytochrome P450 (CYP)-isoforms (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4) or of uptake or efflux transporters (OAT1, OAT3, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, BCRP, P-glycoprotein).
Eflornithine did not induce mRNA expression of CYP1A2, CYP2B6 or CYP3A4 in human hepatocytes.
A study in healthy adults has shown that the consumption of food immediately prior to dosing has no effect on the bioavailability of eflornithine.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
The effects of eflornithine on human fertility are unknown, as no clinical fertility studies were conducted with eflornithine.
In a rat peri-and postnatal reproductive toxicity study, F1 offspring of dams administered eflornithine in the diet at average doses up to 7974 mg/m2/day (approximately 5-fold the clinical exposure of Ifinwil based on body surface area) had a lower fertility index than offspring from control dams.
(Category B3)
There are no clinical data regarding the use of eflornithine during pregnancy. Based on animal data and its mechanism of action (see Section 5.1 Pharmacodynamic Properties), Ifinwil could cause fetal harm if administered during pregnancy. The use of Ifinwil during human pregnancy is not recommended. Verify pregnancy status prior to initiating treatment with Ifinwil, and advise patients of the potential risk to a fetus.
In animal studies, eflornithine demonstrated reproductive toxicity. Oral administration of eflornithine to pregnant rats and rabbits during the period of organogenesis resulted in embryolethality at doses equivalent to the recommended human dose, and as low as one fifth the recommended human dose.
Studies were conducted in rats and rabbits over a range of doses up to 1200 mg/m2/day and 1620 mg/m2/day respectively, covering the period of organogenesis. Maternal toxicity was not observed. Embryofetal toxicity was observed consisting of decreased fetal weights with increased incidence of skeletal variations at 200 mg/kg/day (rats) and early resorption, increased pre-implantation and post-implantation loss, and reduced fetal weights at 135 mg/kg/day (rabbits). No fetal skeletal malformations or external and visceral anomalies were noted in rabbits.
There are no clinical data regarding the use of eflornithine in lactation. It is unknown whether eflornithine is excreted in human milk. A risk to the newborns/infants cannot be excluded. Patients receiving Ifinwil should not breast-feed during treatment and for 1 week after the last dose.
In an oral peri-and postnatal reproductive toxicity study in rat, eflornithine doses greater than 2880 mg/m2/day, during the last week of gestation and continuing to weaning resulted in significantly reduced pup weights continuing throughout the growth period in female pups and for 5-weeks in male pups was performed in rats.
Contraception.
Patients who could become pregnant, or whose sperm could cause their partner to become pregnant, should use effective contraception during treatment with Ifinwil and for 1 week after the last dose.4.7 Effects on Ability to Drive and Use Machines
Treatment with Ifinwil may have a minor influence on the ability to drive and use machines as it can lead to adverse events such as fatigue.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
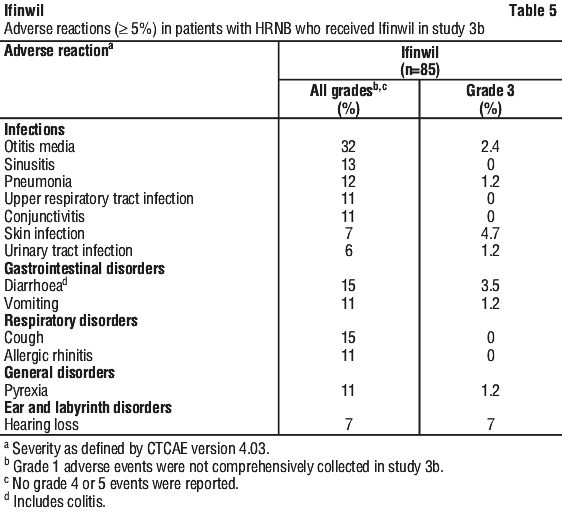
The pooled safety population described in Section 4.4 Special Warnings and Precautions for Use comprises 360 patients with exposure to Ifinwil as a single agent, taken orally at doses ranging from 192 - 768 mg twice daily, based on body surface area (BSA), until disease progression, unacceptable toxicity, or for a maximum of 2 years in patients who demonstrated at least a partial response to prior multiagent, multimodality therapy for newly diagnosed or relapsed/refractory high-risk neuroblastoma in Study 3b (n=101; NCT02395666) and Study 14 (n=259; NCT02679144). The duration of exposure was 6 months or longer for 84% and 12 months or longer for 73% of the pooled safety population. Amongst patients in the pooled safety population, the most common (≥ 5%) adverse reactions were hearing loss (11%), otitis media (10%), pyrexia (7%), pneumonia (5%), and diarrhoea (5%). The most common (≥ 2%) Grade 3 or 4 laboratory abnormalities were increased ALT (11%), increased AST (6%), decreased neutrophils (4.2%), and decreased haemoglobin (3.3%).
Tabulated list of adverse reactions.
The safety of Ifinwil was evaluated in Study 3b (see Section 5.1 Pharmacodynamic Properties, Clinical efficacy). Eligible patients were paediatric patients with high-risk neuroblastoma (HRNB) who demonstrated at least a partial response to prior multiagent, multimodality therapy including induction, consolidation, and anti-GD2 immunotherapy. Patients received Ifinwil as a single agent taken orally at doses ranging from 192 - 768 mg twice daily, based on body surface area (BSA), until disease progression, unacceptable toxicity, or for a maximum of 2 years (N=85). Among patients who received Ifinwil, 93% were exposed for 6 months or longer and 89% were exposed for greater than one year. The median age of patients who received Ifinwil was 4 years (range: 1 to 17); 59% male; 85% White, 7% Black, 1% Asian, 8% Hispanic or Latino; 87% had International Neuroblastoma Staging System Stage 4 disease; 47% had neuroblastoma with known MYCN-amplification. Serious adverse reactions occurred in 12% of patients who received Ifinwil. Serious adverse reactions in > 1 patient included skin infection (3 patients). Permanent discontinuation of Ifinwil due to an adverse reaction occurred in 11% of patients. Adverse reactions which resulted in permanent discontinuation of Ifinwil in > 1 patient included hearing loss. Dose reductions of
Ifinwil due to an adverse reaction occurred in 8% of patients. Adverse reactions which required dose reductions in > 1 patient included hearing loss. The most common (≥ 5%) adverse reactions, including laboratory abnormalities, were otitis media, diarrhoea, cough, sinusitis, pneumonia, upper respiratory tract infection, conjunctivitis, vomiting, pyrexia, allergic rhinitis, decreased neutrophils, increased ALT, increased AST, hearing loss, skin infection, and urinary tract
infection.
Table 5 summarises the adverse reactions in Study 3b.
 Clinically relevant adverse reactions in < 5% of patients included rash, extremity pain, and
alopecia.
Clinically relevant adverse reactions in < 5% of patients included rash, extremity pain, and
alopecia.
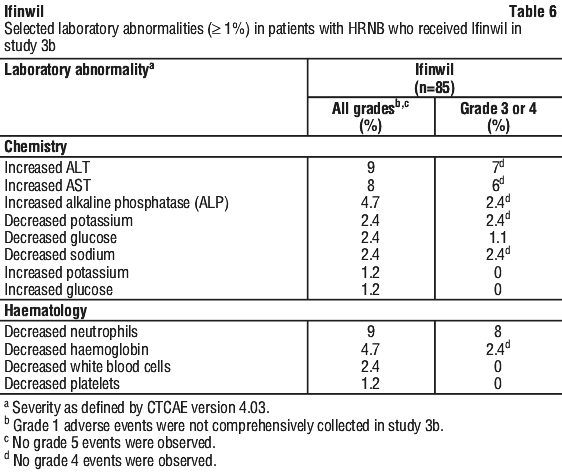
Table 6 summarises the laboratory abnormalities observed in Study 3b.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
No cases of eflornithine overdose have been reported.
In the event of overdose, patients should be carefully observed for signs or symptoms of adverse reactions and general symptomatic and supportive measures should be initiated, as appropriate.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antineoplastic and immunomodulating agents - ATC code L01XX79.
Mechanism of action.
Eflornithine is an irreversible inhibitor of the enzyme ornithine decarboxylase (ODC), the first and rate-limiting enzyme in the biosynthesis of polyamines and a transcriptional target of MYCN. Polyamines are involved in differentiation and proliferation of mammalian cells and are important for neoplastic transformation.
Pharmacodynamic effects.
Inhibition of polyamine synthesis by eflornithine restored the balance of the LIN28/Let-7 metabolic pathway, which is involved in regulation of cancer stem cells and glycolytic metabolism, by decreasing expression of the oncogenic drivers MYCN and LIN28B in MYCN-amplified neuroblastoma. In vitro, eflornithine induced senescence and suppressed neurosphere formation in MYCN-amplified and MYCN non-amplified neuroblastoma cells, indicating a cytostatic effect. Treatment with eflornithine prevented or delayed tumour formation in mice injected with limiting dilutions of MYCN-amplified neuroblastoma cells.
Eflornithine exposure-response relationships and the time course of pharmacodynamic responses are unknown.
Cardiac electrophysiology.
At the recommended dose, Ifinwil did not result in a large mean increase (i.e. > 20 ms) of the QTc interval.
Clinical efficacy.
The efficacy of Ifinwil was demonstrated in an externally controlled trial comparison between Study 3b (investigational arm) and Study ANBL0032 (clinical trial-derived external control arm).
Study 3b.
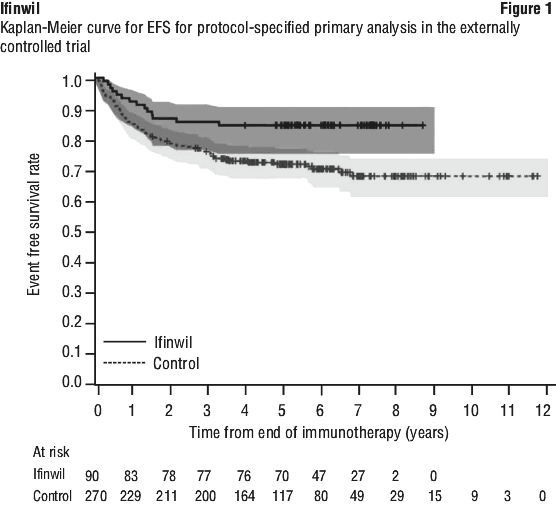
Study 3b (NCT02395666) was a multi-center, open label, non-randomised trial with two cohorts. Eligible patients in one cohort (Stratum 1) were paediatric patients with high-risk neuroblastoma (HRNB) who demonstrated at least a partial response to prior multiagent, multimodality therapy, including induction, consolidation, and anti-GD2 immunotherapy. A total of 105 eligible patients received Ifinwil orally twice daily, dosage based on body surface area (BSA) until disease progression, unacceptable toxicity, or for a maximum of 2 years (see Section 4.2 Dose and Method of Administration). Tumor assessments were performed at 3, 6, 9, 12, 18 months, completion of treatment, and as clinically indicated. Following completion of Ifinwil therapy, patients were followed for a total duration of 7 years. The primary endpoint was event-free survival (EFS), defined as disease progression, relapse, secondary cancer, or death due to any cause. An additional efficacy outcome measure was overall survival (OS), defined as death due to any cause. Study 3b was prospectively designed to compare outcomes to the historical EFS rate from Study ANBL0032 reported in published literature.
External comparator: ANBL0032.
The external control arm was derived from 1,241 patients on the experimental arm of Study ANBL0032, a multi-center, open-label, randomised trial of dinutuximab, granulocyte-macrophage
colony-stimulating factor, interleukin-2, and cis-retinoic acid compared to cis-retinoic acid alone in paediatric patients with HRNB previously treated with induction and consolidation therapy who achieved at least a partial response to prior autologous stem cell transplant. Tumour assessments were performed post-immunotherapy at 3, 6, 9, 12, 18, 24, 30, and 36 months, then per standard of care for a total of 10 years.
Externally controlled trial.
The efficacy population for the comparative analysis of Study 3b and ANBL0032 included patients from both studies who were less than 21 years of age with histologic verification of HRNB and who demonstrated at least a partial response based on imaging, with no evidence of disease in the bone marrow, at the end of immunotherapy, and did not experience an EFS event prior to starting Ifinwil maintenance therapy (for Study 3b), or for at least 30 days from the end of immunotherapy (for ANBL0032). Eligible patients on Study 3b received immunotherapy on ANBL0032 or were treated off study according to the ANBL0032 protocol. Patients who met the criteria for the comparison and had complete data for specified clinical covariates were matched (1:3) using propensity scores; the matched efficacy populations for the primary analysis included 90 patients treated with Ifinwil and 270 control patients from ANBL0032. The demographic characteristics of the primary analysis population (N=360) were 59% male; median age at diagnosis 3 years (range: 0.1 to 20.1); 88% White, 6% Black, 4% Asian, 7% Hispanic. The majority of patients had Stage 4 disease (86%) and MYCN amplification was observed in 44% of tumours. End of immunotherapy responses were complete response (CR; 87%), very good partial response (VGPR; 8%), or partial response (PR; 5%).
In the protocol-specified primary analysis, the EFS hazard ratio (HR) was 0.48 (95% CI: 0.27, 0.85) and OS HR was 0.32 (95% CI: 0.15, 0.70). The Kaplan-Meier plot for the primary analysis of EFS, with shaded bands for each curve representing the point-wise 95% confidence intervals, is shown in Figure 1. Given the uncertainty associated with the externally controlled study design, supplementary analyses in subpopulations or using alternative statistical methods were performed. In these analyses, the EFS HR ranged from 0.43 (95% CI: 0.23, 0.79) to 0.59 (95% CI: 0.28, 1.27), and the OS HR ranged from 0.29 (95% CI: 0.11, 0.72) to 0.45 (95% CI: 0.21, 0.98).
5.2 Pharmacokinetic Properties
Absorption.
Following oral administration to humans, peak plasma concentrations (Cmax) were achieved (Tmax) after approximately 3.5 hours post dose administration.
The Cmax and AUC (area under the concentration-time curve) of eflornithine were not affected by food (high fat and high calories). Administration of crushed tablets in a standard pudding admixture had no effect on eflornithine exposure (Cmax and AUC6 h).
Distribution.
Eflornithine does not specifically bind to human plasma proteins. The volume of distribution (Vz/F) of eflornithine is 24.3L.
Elimination.
Terminal plasma elimination half-life is approximately 3.5 hours. Clearance Cl/F of eflornithine is 5.3 L/h.
In animal studies, 80% of the drug was excreted in urine.
Specific populations.
Pharmacokinetic analyses from patients in Study 14 suggested that age (1 year to 19 years), sex, or body surface area (0.4 m2 to 2 m2), and mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin > 1 x ULN and any AST) had no clinically meaningful effects on eflornithine exposure.
Effect of renal impairment.
After a single oral Ifinwil dose of 576 mg, exposure (AUC) of eflornithine was approximately 2-fold higher in adults with moderate renal impairment and 4-fold higher in adult subjects with severe renal impairment when compared to adult subjects with normal renal function.
5.3 Preclinical Safety Data
Preclinical safety studies were dosed on a mg/kg basis.
Genotoxicity.
Eflornithine was negative for genotoxic activity in the Ames reverse mutation assay, in vitro clastogenic assays and in vivo mouse micronucleus assays.
Carcinogenesis.
No clinically relevant carcinogenicity findings were observed in 104-week oral carcinogenicity studies in mice and rats at doses up to 1000 mg/kg/day and 600 mg/kg/day of eflornithine, respectively. This represents a 2-fold clinical exposure based on BSA.6 Pharmaceutical Particulars
6.1 List of Excipients
Silicified microcrystalline cellulose, pregelatinised maize starch, silicon dioxide, magnesium stearate.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C.
Keep the container tightly closed / airtight.
6.5 Nature and Contents of Container
White HDPE bottle with a foil induction seal, containing a silica gel desiccant and polyester coil with a child resistant cap.
Each bottle contains 100 tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
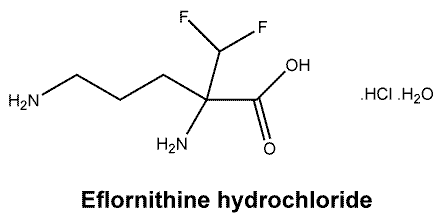
Chemical structure.
C6H15ClF2N2O3.
CAS number.
96020-91-6.7 Medicine Schedule (Poisons Standard)
(S4) Prescription Only Medicine.
Summary Table of Changes
