What is in this leaflet
This leaflet answers some common questions about Ilaris.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine. You can also download the most up to date leaflet from www.novartis.com.au. Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Ilaris against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Ilaris is used for
Ilaris is used to treat certain auto-inflammatory diseases which are collectively known as Cryopyrin-Associated Periodic Syndromes. These include:
- Familial Cold Auto-inflammatory Syndrome (also called Familial Cold Urticaria)
- Muckle-Wells Syndrome
- Neonatal-Onset Multisystem Inflammatory Disease (also called Chronic Infantile Neurological, Cutaneous, Articular Syndrome)
With these conditions, the body produces excessive amounts of a chemical messenger called interleukin-1 beta. This may lead to symptoms such as fever, headache, fatigue, skin rash, painful joints and muscles. In some people, more severe outcomes such as hearing impairment are observed.
Ilaris belongs to a group of medicines called interleukin-1 inhibitors. The active substance in Ilaris is canakinumab.
It binds to interleukin-1 beta, blocking its activity. This leads to an improvement in symptoms.
Ilaris is injected every 8 weeks as a single dose under the skin (also called subcutaneous injection).
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
Ilaris is available only with a doctor's prescription and is not addictive.
It can be used in adults and children aged 4 years and older.
Before you have Ilaris
When you must not take it
Do not take Ilaris if you have an allergy to:
- canakinumab, the active ingredient, or to any of the other ingredients listed at the end of this leaflet.
- any other similar medicines (such as medicines of the same class).
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to have it
Tell your doctor if you have or have had any of the following medical conditions:
- infection or a history of recurring infections, including tuberculosis
- neutropenia, where certain white blood cell counts are low
Tell your doctor if you are pregnant or plan to become pregnant. Ilaris is not recommended for use during pregnancy unless clearly needed. Your doctor can discuss with you the risks and benefits involved.
Tell your doctor if you are breast-feeding. Breastfeeding is not recommended while you are taking Ilaris. It is not known whether Ilaris passes into breast milk and could affect your baby.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes. Your doctor will want to know if you are prone to allergies.
If you have not told your doctor about any of the above, tell them before you start taking Ilaris.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop. Some medicines and Ilaris may interfere with each other. These include:
- vaccinations; you must not be given "live vaccines" while being treated with Ilaris
- medicines called TNF inhibitors (such as etanercept, adalimumab or infliximab).
Such medicines may be affected by Ilaris or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How Ilaris is given
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
Ilaris is intended for subcutaneous use. This means that it is injected through a short needle into the fatty tissue just under the skin. It is injected as a single dose every 8 weeks.
The injection may be given by your doctor or nurse or you may be taught how to inject yourself with the medicine.
If you do not understand the instructions on the label ask your doctor or pharmacist for help.
How much is given
Every 8 weeks a single dose of Ilaris is injected under the skin.
The recommended dose of Ilaris is:
- 150 mg for patients with body weight of more than 40 kg.
- 2 mg/kg for patients with body weight between 15 kg and 40 kg (example: a 25 kg child should receive a 50 mg injection).
- If the rash and other inflammation symptoms have not resolved 7 days after treatment start, your treating physician may consider a second dose of 150 mg (body weight more than 40 kg) or 2mg/kg (body weight between 15 kg and 40 kg). Depending on the effect achieved, your treating physician may decide to increase your regular dose to 300 mg (body weight more than 40 kg) or to 4 mg/kg (body weight between 15 kg and 40 kg) every 8 weeks.
Do not exceed the recommended dose.
Injecting Ilaris yourself
Discuss with your doctor whether or not you will inject Ilaris yourself. After proper training in injection technique, you may inject it yourself.
Do not try to inject yourself if you have not been properly trained or if you are not sure how to do it.
Read the following instructions all the way through before beginning.
Use the following instructions to prepare for the injection:
- Find a clean, comfortable area.
- Wash your hands with soap and water.
- Always use new, unopened needles and syringes. Avoid touching the needles and the tops of the vials.
- Gather together the necessary items included in the pack:
- one vial of Ilaris powder for injection. - Gather together the necessary items not included in the pack:
- one vial of sterile water for injection ("water") (do not refrigerate)
- one 1.0 mL syringe
- one 18 G x 2" (50 mm) needle for reconstituting the powder ("transfer needle")
- one 27 G x 0.5" (13 mm) needle for injecting ("injection needle")
- alcohol swabs
- clean, dry cotton swabs
- an adhesive bandage
- a proper disposal container for used needles, syringe and vials (sharps container).
Use the following list of instructions to make the solution of Ilaris:
- Remove the protective caps from the Ilaris vial and water vial. Do not touch the vial stoppers. Clean the stoppers with the alcohol swab.
- Open the wrappers containing the syringe and the transfer needle (bigger one) and attach the needle to the syringe.
- Carefully remove the cap from the transfer needle and set the cap aside. Pull the plunger all the way down to the 1.0 mL mark, filling the syringe with air. Insert the needle into the water vial through the centre of the rubber stopper.
- Gently push the plunger all the way down until air is injected into the vial.
- Invert the vial and syringe assembly and bring to eye level.
- Make sure the tip of the transfer needle is covered by the water and slowly pull the syringe plunger down to slightly past the 1.0 mL mark. If you see bubbles in the syringe, remove bubbles as instructed by your healthcare provider or pharmacist.
- Make sure 1.0 mL of water is in the syringe, then withdraw the needle from the vial (there will be water remaining in the vial).
- Insert the transfer needle through the centre of the stopper of the vial of Ilaris powder, taking care not to touch the needle or the stopper. Slowly inject 1.0 mL of water in to the vial containing the Ilaris powder.
- Carefully remove the syringe with the transfer needle from the vial and recap the needle as instructed by your healthcare provider or pharmacist.
- Without touching the rubber stopper, swirl (do not shake) the vial slowly at an angle of about 45 degrees for approximately 1 minute. Allow to stand for 5 minutes.
- Gently turn the vial head over tail ten times, again taking care not to touch the rubber stopper.
- Allow to stand for about 5 minutes at room temperature to obtain a clear solution. Do not shake. Do not use if particles are present in the solution.
- Make sure all of the solution is in the bottom of the vial. If drops remain on the stopper, tap the side of the vial to remove them. The solution should be clear and essentially free of visible particles.
- If not used within 1 hour of mixing, the solution should be stored in the refrigerator (2 to 8°C) and used within 24 hours.
Use the following list of instructions to prepare the injection:
- Clean the rubber stopper of the vial containing the Ilaris solution with a new alcohol swab.
- Uncap the transfer needle again. Pull the plunger of the syringe all the way down to the 1.0 mL mark, filling the syringe with air. Insert the syringe needle into the vial of Ilaris solution through the centre of the rubber stopper. Gently push the plunger all the way down until air is injected into the vial. Do not inject air into the medication.
- Do not invert the vial and syringe assembly. Insert the needle all the way into the vial until it reaches the bottom edge.
- Tip the vial to ensure that the required amount of solution can be drawn into the syringe.
The required amount depends on the dose to be administered (0.2 mL to 1.0 mL). Your healthcare provider will instruct you on the right amount for you. - Slowly pull the syringe plunger up to the correct mark (0.2 to 1.0 mL), filling the syringe with Ilaris solution. If there are air bubbles in the syringe, remove bubbles as instructed by your healthcare provider. Ensure that the correct amount of solution is in the syringe.
- Remove the syringe and needle from the vial. (There may be solution remaining in the vial.) Recap the transfer needle as instructed by your healthcare provider or pharmacist.
- Remove the transfer needle from the syringe. Place the transfer needle in the sharps container.
Use the following list of instructions to give the injection:
- Open the wrapper containing the injection needle and attach the needle to the syringe. Set the syringe aside.
- Choose an injection site on the upper arm, upper thigh, abdomen or buttocks. Do not use an area that has a rash or broken skin, or is bruised or lumpy. Avoid injecting into scar-tissue as this may lead to insufficient exposure to canakinumab. Avoid injecting into a vein.
- Clean the injection site with a new alcohol swab. Allow the area to dry. Uncap the injection needle.
- Gently pinch the skin up at the injection site. Hold the syringe at a 90-degree angle and in a single, smooth motion, push the needle straight down completely into the skin.
- Keep the needle all the way in the skin while slowly pushing the syringe plunger down until the barrel is empty. Release the pinched skin and pull the needle straight out. Dispose of the needle and syringe in the sharps container without recapping or removing the needle.
- Do not rub the injection area. If bleeding occurs, apply a clean, dry cotton swab over the area, and press gently for 1 to 2 minutes, or until bleeding stops. Then apply an adhesive bandage.
How long to take it
Keep taking this medicine for as long as your doctor tells you.
If you forget to have it
If you forget to inject it, inject the next dose as soon as you remember then contact your doctor to discuss when you should take the next dose. You should continue with injections at 8-week intervals, as before.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to have your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Ilaris. Do this even if there are no signs of discomfort or poisoning.
While you are taking Ilaris
Things you must do
Keep all of your doctor's appointments so that your progress can be checked. Your doctor will do tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Tell your doctor if you have a temperature or chills, or another sign of an infection. You may need medical treatment.
Tell your doctor if you experience any signs of an allergic reaction such as difficulty breathing or swallowing, nausea, dizziness, skin rash, itching, hives or palpitations.
If you want to be vaccinated, tell your doctor you are taking Ilaris before you have the vaccination. Some vaccines may not be suitable for you.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Ilaris.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
Things you must not do
Do not take Ilaris to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Ilaris. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following which might worry you:
- redness, pain or itching at the site of injection
- combination of sore throat, runny nose, blocked nose, sneezing with or without fever
- dizziness or spinning sensation.
If any of the following happen, tell your doctor immediately:
- constant "flu-like" symptoms such as fever, chills, sore throat, aching joints, swollen glands, cough, or any other signs of infection such as infection of a cut or scratch
- signs of an allergy such as rash, itching or hives on the skin; swelling of the face, lips, tongue or other part of the body; shortness of breath, wheezing or difficulty breathing or swallowing, nausea, dizziness, palpitations or low blood pressure
- burning sensation on urination or increased urgency to urinate.
The above list includes serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed here or not yet known may happen in some people. Some of these side effects can only be found by laboratory testing.
After taking Ilaris
Storage
If you have to store Ilaris:
- Keep it in a refrigerator (2°C to 8°C). Do not freeze it.
- Keep the vial in the outer carton in order to protect it from light.
- Do not store Ilaris or any other medicine in the bathroom or near a sink.
- Do not leave it in the car or on a window sill.
Keep the medicine where young children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Ilaris is supplied as a powder for injection in a single-use glass vial.
The powder is white. It can be in a whole or a fragmented cake.
Each pack contains one or four single-dose vials.
Ingredients
Ilaris powder for injection vials contain 150 mg canakinumab as the active ingredient. They also contain:
- sucrose
- histidine
- histidine hydrochloride monohydrate
- polysorbate 80
- dilute hydrochloric acid.
This medicine does not contain gluten, tartrazine or any other azo dyes.
Sponsor
Ilaris is supplied in Australia by:
Novartis Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
North Ryde NSW 2113
Telephone 1 800 671 203
®= Registered Trademark
This leaflet was prepared in
March 2012.
Australian Registration Numbers:
AUST R 159573 - powder for injection vial only
AUST R 187078 - powder for injection vial and water for injections diluent vial
Published by MIMS July 2025
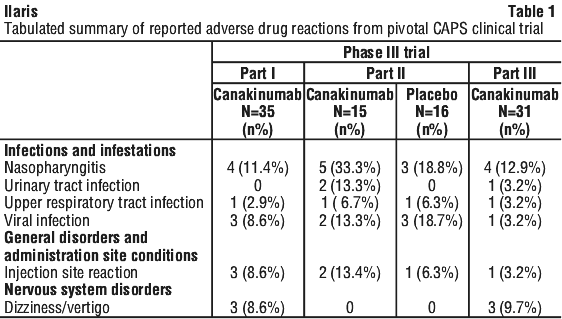 In the long-term, open label studies with dose-escalation, events of infections (gastroenteritis, respiratory tract infection, and upper respiratory tract infection), vomiting and dizziness were more frequently reported in the 600 mg or 8 mg/kg dose group than in other dose groups.
In the long-term, open label studies with dose-escalation, events of infections (gastroenteritis, respiratory tract infection, and upper respiratory tract infection), vomiting and dizziness were more frequently reported in the 600 mg or 8 mg/kg dose group than in other dose groups.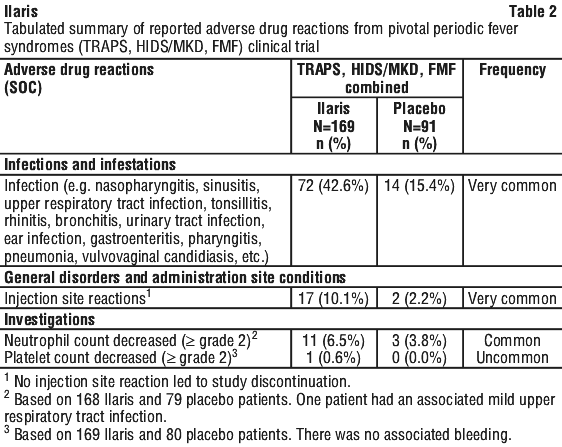
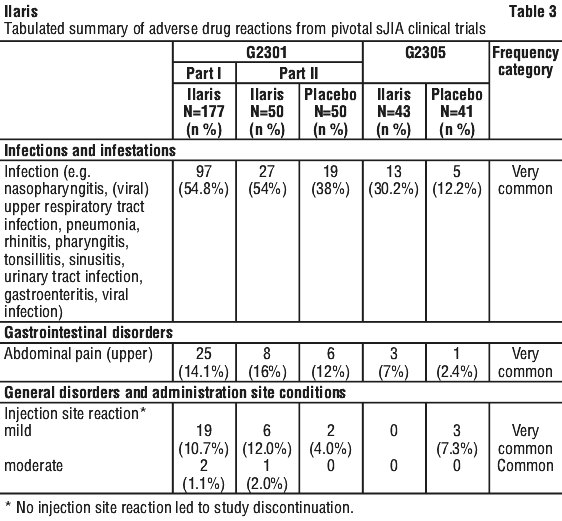
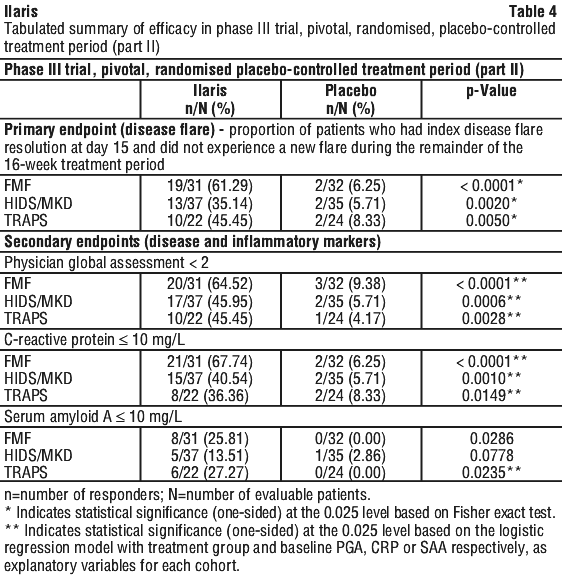
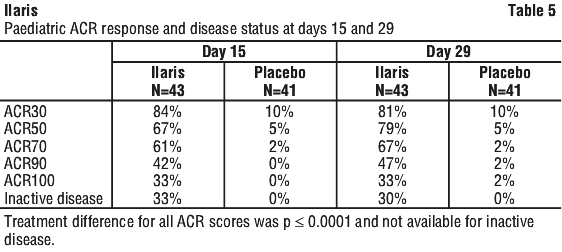 Ilaris treatment improved components of paediatric ACR core set as compared to placebo at Days 15 and 29 (Table 6). All patients treated with Ilaris had no fever at day 3 compared to 86.8% of patients treated with placebo (p=0.0098).
Ilaris treatment improved components of paediatric ACR core set as compared to placebo at Days 15 and 29 (Table 6). All patients treated with Ilaris had no fever at day 3 compared to 86.8% of patients treated with placebo (p=0.0098).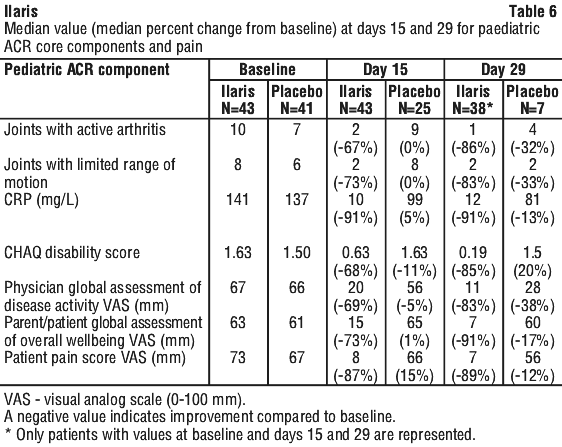 No patient in Study G2305 discontinued because of an adverse event.
No patient in Study G2305 discontinued because of an adverse event.