What is in this leaflet
This leaflet answers some common questions about ILUMYA (pronounced e-loom-e-a).
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking ILUMYA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist. Keep this leaflet with the medicine, you may need to read it again.
What ILUMYA is used for
ILUMYA is used to treat a skin condition called plaque psoriasis in adults with moderate to severe disease who are candidates for systemic therapy. Plaque psoriasis causes inflammation of the skin. ILUMYA reduces the inflammation and other symptoms of the disease, and promotes skin clearance.
ILUMYA contains the active substance tildrakizumab.
Tildrakizumab is a humanized monoclonal antibody and belongs to a group of medicines called interleukin (IL) inhibitors. This medicine works by neutralising the activity of a protein called IL-23, a substance found in the body which is involved in normal inflammatory and immune responses. In patients with plaque psoriasis, the body produces increased amounts of this protein. This may lead to symptoms such as itching, pain and scaling.
Your doctor may have prescribed it for another reason.
ILUMYA has not been studied in patients under 18 years of age.
ILUMYA may be used in elderly patients aged 65 years and over.
ILUMYA is available only with a doctor’s prescription. It is not addictive.
If you have any questions about how ILUMYA works or why this medicine has been prescribed for you, ask your doctor, pharmacist or nurse.
Before you take ILUMYA
Tell your doctor if you have any of the conditions mentioned below or if you have ever experienced any of these conditions.
When you must not take it
Do not take ILUMYA If:
- you have had a severe allergic reaction to tildrakizumab or any of the other ingredients listed at the end of this leaflet. (see Product description)
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
If you think you may be allergic, ask your doctor for advice before using ILUMYA. - you have an active infection which your doctor thinks is important.
- the product appears cloudy, discolored or contains particles.
- the expiry date on the pack has passed or if packaging is torn or shows signs of tampering. Return expired or damaged stock to your pharmacist.
- you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor before taking ILUMYA if:
- you currently have an infection or if you have long-term or repeated infections.
- you have tuberculosis or have been in contact recently with someone who has.
- you had a recent vaccination or someone close to you has had a vaccination or if you will receive a live vaccine during treatment with ILUMYA. Live vaccines such as oral polio vaccine, should not be given while receiving ILUMYA.
- You have or have had any type of cancer.
- you are pregnant, think that you may be pregnant or are planning to have a baby.
- The effects of ILUMYA in pregnancy are unknown. It is preferable to avoid the use of ILUMYA in pregnancy.
If you are a woman of childbearing potential, you are advised to avoid becoming pregnant and must use an effective method of contraception whilst having treatment with ILUMYA and for at least 17 weeks after treatment.
Talk to your doctor about the benefits to you using ILUMYA verses the potential risk to your unborn child. - you are breast-feeding or plan to breast-feed.
If you have not told your doctor about any of the above, tell them before you start taking ILUMYA.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including:
- any that you get without a prescription from your pharmacy, supermarket or health food shop.
- If you have had or are due to have a vaccination. You should not receive certain types of vaccines (live vaccines) while taking ILUMYA.
How ILUMYA is given
ILUMYA may be given to you by your doctor or nurse, or you or a caregiver may be taught how to inject yourself with the medicine.
You and your doctor, nurse or pharmacist should decide if you should inject ILUMYA yourself. Do not inject yourself if you have not been properly trained by your doctor, nurse or pharmacist. A caregiver may also give you your ILUMYA injection after proper training.
How much is given
Your doctor will decide how much ILUMYA you need. The usual dose of ILUMYA is 100mg given by subcutaneous injection.
After the first dose you will need to take the same dose 4 weeks later and then all future doses once every 12 weeks.
If your doctor gives you different dosing instructions follow your doctors instructions.
Inject the whole content of the syringe.
Do not shake the syringe.
Do not exceed the recommended dose.
Injecting ILUMYA yourself
Follow all directions given to you by your doctor, nurse or pharmacist carefully, if they differ from these instructions follow your healthcare professional’s instructions.
If you do not understand the instructions, ask your doctor, nurse or pharmacist for help.
ILUMYA is intended for subcutaneous use. This means that it is injected into the fatty tissue just under the skin.
How to use the pre-filled syringe
Read all of the instructions on how to use the pre-filled syringe before beginning the injection.
Keep the instructions and refer to them as needed. These instructions will help you to inject correctly using the pre-filled syringe.

Preparation
- For a more comfortable injection, take the carton containing the ILUMYA pre-filled syringe out of the refrigerator and let it sit at room temperature for 30 minutes before injecting.

- Check the expiration date on the carton and pre-filled syringe and discard if the date has passed.
- Assemble other necessary supplies, not included in the ILUMYA pack:
- an alcohol swab,
- a cotton ball or gauze,
- and a sharps container or similar puncture proof container of hard plastic or glass, for syringe disposal. - Remove the pre-filled syringe from the carton when ready to inject.
DO NOT pull off the needle cover until you are ready to inject. - Inspect ILUMYA visually and discard if it contains particles, is cloudy or discoloured, or if the syringe is damaged.
- ILUMYA is a clear to slightly opalescent (milky) and colorless to slightly yellow, solution.
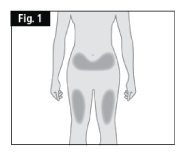
- Air bubbles may be present; there is no need to remove them. - Choose an injection site with clear skin and easy access such as front of thighs, or lower abdomen, but not the area five centimetres around the navel (belly button) (Fig. 1).
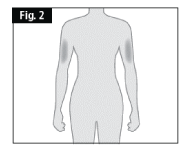
If a caregiver, or healthcare professional is giving you the injection, the outer upper arms may also be used (Fig. 2).
- DO NOT administer where the skin is tender, bruised, abnormally red, hard or affected by psoriasis.
- DO NOT inject into scars, stretch marks, or blood vessels.
Giving the Injection
Your Doctor, Pharmacist or Nurse will give you instructions on how to inject ILUMYA subcutaneously and their instructions might differ from these instructions.
- When you are ready to inject, wash your hands thoroughly with soap and water.
- Find a clean, comfortable area to give the injection.
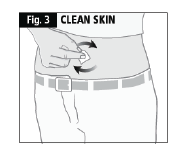
- Using a circular motion, clean the injection site with the alcohol swab. Leave to dry and do not touch the cleaned area again before injecting (Fig. 3).
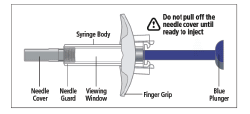
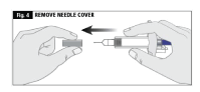
- While holding the body of the syringe, remove the needle cover as shown and discard (Fig. 4).
- To inject:
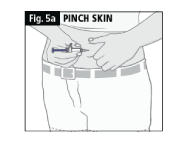
a. Gently press your skin between your thumb and index finger (Fig. 5a).
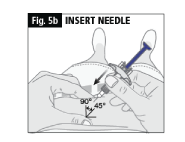
b. Insert the needle in to the skin at a 45-90 degree angle (Fig. 5b).
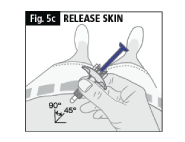
c. Release the skin before you push on the plunger (Fig. 5c).
d. Slowly press down the blue plunger with your thumb as far as it will go (Fig. 5d). (This will activate the safety mechanism that ensures full retraction of the needle after the injection is given.)
The whole of the Dose Window will be blue when the injection is complete
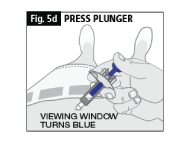
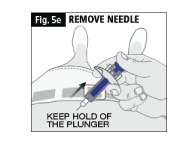
e. DO NOT LET GO OF THE PLUNGER until you have removed the needle from the skin (Fig. 5e).
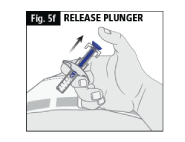
f. When you release the plunger the safety lock will draw the needle inside the needle guard (Fig. 5f).
g. There may be a small amount of blood at the injection site. You can press a cotton ball or gauze over the injection site and hold it for 10 seconds.
Do not rub the injection site.
You may cover the injection site with a small adhesive bandage, if needed.
- Dispose of the syringe in a sharps disposal container right away after use (Fig. 6).

Never try to reuse the pre-filled syringe.
How long to take ILUMYA
ILUMYA is for long term treatment. Your doctor will regularly monitor your condition to check that the treatment is having the desired effect.
Do not stop taking ILUMYA just because you feel better. It is important that you do not stop unless your doctor tells you to. It is not dangerous to stop taking ILUMYA but if you stop your psoriasis symptoms may come back.
If you use more ILUMYA than you should (overdose)
If you accidentally inject more ILUMYA than you should or you take a dose sooner than according to the doctor’s prescription, immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital.
Do this even if there are no obvious signs of discomfort or poisoning.
If you miss a dose
If you have forgotten to inject a dose of ILUMYA, inject the next dose as soon as you remember. Then talk to your doctor to discuss when you should inject the next dose.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
While you are taking ILUMYA
Things you must do
Keep all of your doctor’s appointments so that your progress can be checked.
Look out for infections and allergic reactions
ILUMYA can potentially cause serious side effects, including infections and allergic reactions. You must look out for signs of these conditions while you are taking ILUMYA.
Stop using ILUMYA and tell your doctor or seek medical help immediately if you notice any signs indicating a possible serious infection or an allergic reaction (see Side effects).
If you need to be vaccinated, tell your doctor you are taking ILUMYA before you have the vaccination.
You should not be given a live vaccination while being treated with ILUMYA.
As soon as you start taking ILUMYA tell all doctors, dentists and pharmacists who are treating you that you are taking ILUMYA.
Remind your doctor or pharmacist you are taking ILUMYA before you start any new medicine.
Tell your doctor if you become pregnant while taking ILUMYA.
Tell your doctor or pharmacists as soon as possible if you do not feel well, you think you have an infection or you have any unwanted side effects.
Things you must not do
Never leave the pre-filled syringe lying around where others might tamper with it.
Do not use this medicine if the liquid contains easily visible particles, is cloudy or is distinctly brown.
Do not use the pre-filled syringe if any of the outer packaging is damaged, the expiry date has passed or the seals are broken.
It may not be safe for you to use.
Do not shake the pre-filled syringe.
Do not use the syringe if it is damaged or cap has been removed.
Do not take it to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Things to be careful of
Dispose of the used ILUMYA pre-filled syringe immediately after use in a sharps container.
The ILUMYA pre-filled syringe should never be re-used.
If you stop using ILUMYA
Do not stop taking your medicine or change dosage without checking with your doctor first.
Side effects
Tell your doctor, pharmacist or nurse as soon as possible if you do not feel well while taking ILUMYA.
As with all medicines, patients treated with ILUMYA may experience side effects, although not everybody gets them.
All medicines can have side effects. Sometimes they are serious, most of the time they are not.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Some side effects are common.
- Upper respiratory tract infections with symptoms such as sore throat and stuffy nose (nasopharyngitis)
- Headache
- Injection site pain
Stop using ILUMYA and tell your doctor immediately or go to hospital as you may need urgent medical attention if you have an allergic reaction.
Symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Your doctor will decide if and when you may restart treatments.
Ask your doctor, nurse or pharmacist to answer any questions you may have.
Other side effects not listed above may also occur in some people. Tell your doctor if you notice anything else that is making you feel unwell.
Many medicines like ILUMYA that may decrease the activity of your immune system, may increase the risk of cancer. Tell your doctor if you notice anything unusual or if you are concerned about any aspect of your health.
After taking ILUMYA
Storage
In general:
ILUMYA pre-filled syringe should be stored in a refrigerator (between 2°C and 8°C), out of the sight and reach of children.
If required:
For purposes of travel or transporting between refrigerated storage, ILUMYA can be stored for up to 30 days at or below 25°C. After 30 days out of the refrigerator, the ILUMYA prefilled syringe should be immediately used or discarded.
Keep the product in the original carton. Protect from light until the time of use.
Do not store ILUMYA or any other medicine in the bathroom, near a sink, or leave it in the car or on a window sill.
Do not shake.
Do not freeze.
Disposal
If your doctor tells you to stop using ILUMYA or you find your pre-filled syringe has passed its expiry date, please return it to your pharmacist.
Empty pre-filled syringes should be disposed of in a sharps container or similar puncture proof container composed of hard plastic or glass.
Ask your doctor, nurse or pharmacist where you can dispose of the container once it is full.
Product description
What it looks like
ILUMYA solution for injection is a clear to slightly opalescent (milky). The colour may vary from colourless to slightly yellow solution. It is supplied in a carton pack containing 1 single use pre-filled syringe.
Ingredients
Each ILUMYA pre-filled syringe contains 100 mg tildrakizumab as the active substance.
The other ingredients are: L-histidine, L-histidine hydrochloride monohydrate, polysorbate 80, sucrose and water for injection.
This medicine does not contain lactose, gluten, tartrazine or any other azo dyes.
Sponsor
ILUMYA is supplied in Australia by:
Sun Pharma ANZ Pty Ltd
ABN 17 110 871 826
Macquarie Park Sydney NSW 2113
Telephone 1 800 726 229
Australian Registration Number: 290683
This leaflet was prepared in Sep 2024.
Published by MIMS November 2024
 Through up to 5 years, 525 subjects in reSURFACE 1 (100 mg: 256; 200 mg: 269) and 730 subjects in reSURFACE 2 (100 mg: 381; 200 mg: 349) were exposed to Ilumya. The safety profile of Ilumya was consistent with that observed in the placebo-controlled periods. No meaningful differences were observed in the overall frequencies of drug-related AEs between the 100 mg and 200 mg treatment groups.
Through up to 5 years, 525 subjects in reSURFACE 1 (100 mg: 256; 200 mg: 269) and 730 subjects in reSURFACE 2 (100 mg: 381; 200 mg: 349) were exposed to Ilumya. The safety profile of Ilumya was consistent with that observed in the placebo-controlled periods. No meaningful differences were observed in the overall frequencies of drug-related AEs between the 100 mg and 200 mg treatment groups.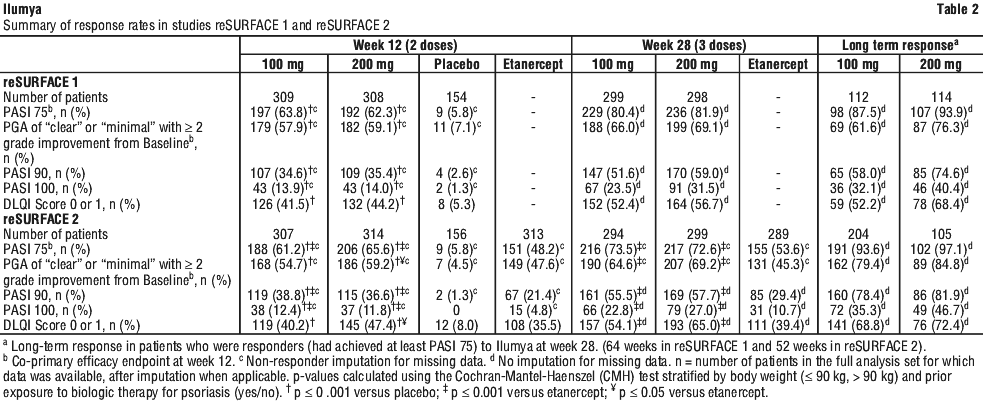
 Both the groups were assessed for safety; no new safety signals were identified.
Both the groups were assessed for safety; no new safety signals were identified.
