



What is in this leaflet
This leaflet answers some common questions about IMBRUVICA capsules and tablets. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given IMBRUVICA against the benefits this medicine is expected to have for you.
If you have any concerns about being given IMBRUVICA ask your doctor or healthcare professional.
Keep this leaflet while you are taking IMBRUVICA. You may need to read it again.
What IMBRUVICA is used for
IMBRUVICA is an anticancer medicine that contains the active substance ibrutinib.
IMBRUVICA is used to treat the following blood cancers in adults:
- Mantle Cell Lymphoma (MCL), a type of cancer affecting the lymph nodes;
- Chronic Lymphocytic Leukaemia (CLL), including Small Lymphocytic Lymphoma (SLL), a type of cancer affecting a type of white blood cell called lymphocytes that also involve the lymph nodes.
- Waldenström's macroglobulinemia (WM), a very rare cancer affecting the lymphocytes
IMBRUVICA works by blocking a protein in the body that helps cancer cells live and grow. This protein is called Bruton's tyrosine kinase. By blocking this protein, IMBRUVICA may help kill and reduce the number of cancer cells and may also slow the spread of the cancer.
Ask your doctor or healthcare professional if you have any questions about why IMBRUVICA has been prescribed for you.
This medicine is available only with a doctor's prescription.
Before you take IMBRUVICA
When you must not use it:
Do not take IMBRUVICA:
- if you are allergic (hypersensitive) to ibrutinib, or other ingredients of IMBRUVICA. See Product Description at the end of this leaflet for a list of ingredients.
Do not take IMBRUVICA:
- if the packaging is torn or shows signs of tampering.
- if the expiry date (month and year) printed on the pack has passed. If you take IMBRUVICA after the expiry date it may not work.
Do not use preparations containing St John's Wort while you are taking IMBRUVICA.
Do not fall pregnant while you are taking IMBRUVICA. If you are pregnant, think you may be pregnant or are planning to have a baby, ask your doctor or healthcare professional for advice before taking IMBRUVICA.
- IMBRUVICA should not be used during pregnancy.
- There is no information about the safety of IMBRUVICA in pregnant women.
- Women of childbearing age must use an effective method of birth control during and up to three months after receiving IMBRUVICA to avoid becoming pregnant while being treated with IMBRUVICA. The time period following treatment with IMBRUVICA where it is safe to become pregnant is not known.
- Tell your doctor immediately if you become pregnant.
Do not breast feed while you are taking IMBRUVICA.
Do not father a child while taking IMBRUVICA and for 3 months after stopping treatment.
- Use condoms and do not donate sperm during treatment and for 3 months after your treatment has finished. If you plan to father a child, talk to your doctor or healthcare professional before taking IMBRUVICA.
IMBRUVICA should not be used by anyone under 18 years of age because it has not been studied in this age group.
Before you start to use it:
Tell your doctor if you have or have had any medical conditions, especially the following:
- if you have ever had unusual bruising or bleeding or are on any medicines or supplements that increase your risk of bleeding
- if you have a history of high blood pressure, irregular heart beat (atrial fibrillation, ventricular tachyarrhythmia) or severe heart failure, or if you feel any of the following: lightheadedness, dizziness, shortness of breath, chest discomfort, swollen legs, or you faint
- if you have liver or kidney problems
- if you have or have had Hepatitis B infection
- if you have recently had any surgery, especially if this might affect how you absorb food or medicines from your stomach or gut
- if you are planning to have any surgery - your doctor may ask you to stop taking IMBRUVICA for a short time.
Taking other medicines:
IMBRUVICA may make you bleed more easily. Tell your doctor if you take other medicine that increase your risk of bleeding. These include the following medicines:
- warfarin, heparin or other medicines to prevent blood clots.
- aspirin and non-steroidal anti-inflammatories (NSAIDS) such as ibuprofen or naproxen
- fish oil and supplements containing vitamin E
The effect of IMBRUVICA or other medicines may be influenced when taking IMBRUVICA with some other medicines.
Tell your doctor if you take any of the following medicines:
- medicines called antibiotics to treat bacterial infections - clarithromycin, telithromycin, ciprofloxacin, erythromycin, rifampin or azithromycin.
- medicines for fungal infections - ketoconazole, posaconazole, itraconazole, fluconazole or voriconazole
- medicines for HIV infection - ritonavir, cobicistat, indinavir, nelfinavir, saquinavir, amprenavir, atazanavir, darunavir/ritonavir or fosamprenavir
- aprepitant - medicine to prevent nausea and vomiting associated with chemotherapy
- nefazodone - medicine for depression
- medicines called kinase inhibitors for treatment of other cancers - crizotinib, imatinib
- medicines called calcium channel blockers for high blood pressure or chest pain - diltiazem, verapamil
- medicines used to treat or prevent irregular heartbeat - amiodarone, dronedarone
- fluvoxamine - medicine used to treat obsessive compulsive disorder
- medicines to prevent seizures or to treat epilepsy or medicines to treat a painful condition of the face called trigeminal neuralgia - carbamazepine, phenytoin
- medicines called statins to treat high cholesterol - rosuvastatin
- St. John's Wort - herbal medicine used for depression
If you are taking digoxin, a medicine used for heart problems, or methotrexate, a medicine used to treat other cancers and to reduce the activity of the immune system (e.g., for rheumatoid arthritis or psoriasis), it should be taken at least 6 hours before or after IMBRUVICA.
IMBRUVICA might interact with other medicines. This may result in greater or lesser effects or even side effects from these medicines.
Tell your doctor if you are taking any other medicines, including medicines you can buy without a prescription from a pharmacy, supermarket or health food shop.
Your doctor can tell you whether you can continue the medicines you are taking or reduce the dose.
Taking IMBRUVICA
Always take IMBRUVICA exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
Effects on the heart
Treatment with IMBRUVICA may affect the heart, especially if you already have heart diseases such as rhythm problems, heart failure, high blood pressure or have diabetes. The effects may be severe and could cause death, including sometimes sudden death. Your heart function will be checked before and during treatment with IMBRUVICA. Tell your doctor immediately if you feel breathless, have difficulty breathing when lying down, swelling of the feet, ankles or legs and weakness/tiredness during treatment with IMBRUVICA – these may be signs of heart failure.
Tests and check-ups before and during treatment
Laboratory tests may show that your blood count contains more white blood cells (called "lymphocytes"), in the first few weeks of treatment. This is expected and may last for a few months. This does not necessarily mean that your blood cancer is getting worse. Your doctor will check your blood counts before or during the treatment and in rare cases they may need to give you another medicine. Talk to your doctor about what your test results mean.
How much IMBRUVICA to take:
The recommended dose of IMBRUVICA for:
- MCL is 560 mg once a day.
- WM and CLL/SLL is 420 mg once a day.
Follow your doctor's instructions on the dose appropriate for you.
Instructions:
- Do not take IMBRUVICA with grapefruit or Seville oranges - this includes eating them, drinking the juice, or taking supplements that might contain them. This is because they can increase the amount of IMBRUVICA in your blood.
- Swallow IMBRUVICA capsules whole with a glass of water. Do not open, break, or chew them.
- Swallow IMBRUVICA tablets whole with a glass of water. Do not break or chew them.
- Try to take IMBRUVICA at the same time each day.
How long to take
Take IMBRUVICA exactly as prescribed by your doctor or healthcare professional. Do not change your dose or stop taking IMBRUVICA until your doctor tells you to.
What do I do if I forget to take IMBRUVICA?
- If it is more than 12 hours until your next dose, take the missed dose as soon as possible. Then continue taking IMBRUVICA at the usual scheduled time.
- If it is less than 12 hours until your next dose, skip the missed dose. Then take the next dose of IMBRUVICA at the usual scheduled time.
- Do not take extra capsules or tablets to make up the missed dose.
If you are not sure what to do, contact your doctor or pharmacist.
What do I do if I take too much? (overdose):
If you think you or anybody else has taken too much IMBRUVICA, contact your doctor, pharmacist or the Poisons Information Centre who will advise you what to do.
You can contact the Poisons Information Centre by dialling:
- Australia: 13 11 26
- New Zealand: 0800 POISON or 0800 764 766.
While you are taking IMBRUVICA
Things you must do:
Be sure to keep all your doctor's appointments so your progress can be checked.
Your doctor may order certain tests, including blood tests, from time to time to make sure the medicine is working and to prevent unwanted side effects.
Be sure to follow up your doctor's instructions about other medicines you should take, and other things you should do.
Tell any other doctors and pharmacists who are treating you that you are taking IMBRUVICA.
If you are about to be started on any new medicines, tell your doctor or pharmacist that you are taking IMBRUVICA.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
Things to be careful of
Driving and using machines
You may feel tired or dizzy after taking IMBRUVICA, which may affect your ability to drive or use any tools or machinery.
Side Effects
Like all medicines, IMBRUVICA can cause side effects, although not everybody gets them. The following side effects may happen with this medicine:
- Bleeding: You may experience bruising or nosebleeds during treatment with IMBRUVICA. Bleeding in the eye or serious internal bleeding, such as bleeding in your stomach, intestine, or brain may occur. Call your doctor or healthcare professional if you have signs or symptoms of serious bleeding, such as blood in your stools or urine or bleeding that lasts for a long time or that you cannot control.
- Leukostasis: You may experience an increase in the number of white blood cells, specifically lymphocytes in your blood. In rare cases, this increase may be severe, causing cells to clump together. Your doctor will monitor your blood counts.
- Infections: You may experience viral, bacterial, or fungal infections during treatment with IMBRUVICA. Contact your doctor if you have fever, chills, body aches, cold or flu symptoms, feel tired or feel short of breath, yellowing of the skin or eyes (jaundice), confusion - these could be signs of an infection.
- Decrease in blood cell counts: Use of IMBRUVICA may cause you to have a low number of red blood cells (anaemia), a low number of neutrophils a type of white blood cell (neutropenia) or a low number of platelets a type cell that help blood to clot (thrombocytopenia). Your doctor or healthcare professional should check your blood counts regularly.
- Interstitial lung disease (ILD): Inflammation within the lungs that may lead to permanent damage has happened with IMBRUVICA treatment. Contact your doctor if you have difficulty breathing or have a persistent cough.
- Heart problems: Irregular heart beat (atrial fibrillation, ventricular tachyarrhythmia) and high blood pressure have occurred with IMBRUVICA treatment. Tell your doctor or healthcare professional if you have any heart problems like chest discomfort, shortness of breath or palpitations.
Heart failure has also been reported. Tell your doctor immediately if you notice breathlessness, difficulty breathing when lying down, swelling of the feet, ankles or legs and weakness/tiredness during treatment with IMBRUVICA. - Tumour lysis syndrome (TLS): Unusual levels of chemicals in the blood caused by the fast breakdown of cancer cells have happened during treatment of cancer and sometimes even without treatment. This may lead to changes in kidney function, abnormal heartbeat, or seizures. Your doctor or another healthcare provider may do blood tests to check for TLS.
- Other cancers: New cancers have occurred in people taking IMBRUVICA, including skin cancer and other cancers.
- Liver problems: Very rarely patients may experience changes in their liver function. Your doctor will monitor your liver function by periodic blood tests. If you notice signs of jaundice such as yellowing of the whites of the eyes please call your doctor immediately.
- Stroke: Temporary or permanent decrease of brain or nerve function due to reduced blood flow to the brain (mini-stroke or stroke).
- Haemophagocytic lymphohistiocytosis: There have been rare reports of excessive activation of white blood cells associated with inflammation, which can be fatal if not diagnosed and treated early. If you experience multiple symptoms such as fever, swollen glands, bruising, or skin rash, contact your doctor immediately.
- Rupture of spleen: Cases of splenic rupture have been reported after stopping IMBRUVICA treatment. Tell your doctor immediately if you develop left upper belly (abdominal) pain, pain below the rib cage or at the tip of your left shoulder.
The most common side effects seen include: diarrhoea; feeling very tired; nausea; headache; swollen hands, ankles or feet; being short of breath; dizziness; fainting; constipation; infected nose, sinuses or throat (cold); fever; vomiting; decreased appetite; low number of a neutrophils (neutropenia); low number of platelets (thrombocytopenia) and low number of red blood cells (anaemia); bruises; skin rash; muscle and joint pain; muscle spasms; low blood sodium levels; urinary tract infection; high blood pressure; sore or inflamed mouth; blurred vision; sore stomach, high levels of uric acid and increased creatine in your blood.
If you have diarrhoea that lasts for more than a week, your doctor may need to give you a fluid and salt replacement or another medicine.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Other side effects not listed above may also occur in some patients. Tell your doctor if you notice anything else that is making you feel unwell.
Product Description
Storage
Store below 30°C. Keep capsules or tablets in the original container.
Do not store it or any medicines in the bathroom or near a sink. Heat and dampness can destroy some medicines.
Keep this medicine out of the sight and reach of children. A locked cupboard at least one-and -a-half metres above the ground is a good place to store medicines.
Do not use this medicine after the expiry date which is stated on the package after EXP.
What it looks like:
Capsules
The hard capsules are white opaque, with "ibr 140 mg" printed in black ink.
IMBRUVICA capsules are supplied in bottles containing 90 or 120 capsules. Not all pack sizes may be marketed.
Tablets
140 mg tablets are yellow-green to green, round, debossed with “ibr” on one side and “140” on the other.
280 mg tablets are purple, oblong-shaped, debossed with “ibr” on one side and “280” on the other.
420 mg tablets are yellow-green to green, oblong-shaped debossed with “ibr” on one side and “420” on the other.
560 mg tablets are yellow to orange, oblong-shaped, debossed with “ibr” on one side and “560” on the other.
IMBRUVICA 140 mg, 280 mg, 420 mg and 560 mg tablets are supplied in cartons containing 30 tablets (3 cardboard wallets containing 10 filmcoated tablets each).
Not all pack sizes may be marketed
Ingredients
Capsules
Active ingredient:
- ibrutinib
Each hard capsule contains 140 mg of ibrutinib.
Other ingredients:
- croscarmellose sodium
- microcrystalline cellulose
- sodium lauryl sulphate
- magnesium stearate
- gelatin
- titanium dioxide (E171)
- black ink
Tablets
Active ingredient:
- ibrutinib
Each tablet contains 140 mg, 280 mg, 420 mg or 560 mg of ibrutinib.
Other ingredients:
- colloidal anhydrous silica
- croscarmellose sodium
- lactose monohydrate
- magnesium stearate
- microcrystalline cellulose
- povidone
- sodium lauryl sulfate
The film-coating contains:
- iron oxide black (140 mg, 280 mg and 420 mg tablets)
- polyvinyl alcohol
- macrogol
- iron oxide red (280 mg and 560 mg tablets)
- talc
- titanium dioxide
- iron oxide yellow (140 mg, 420 mg and 560 mg tablets)
Sponsor
JANSSEN-CILAG Pty Ltd
1-5 Khartoum Rd
Macquarie Park
NSW 2113 Australia
Telephone: 1800 226 334
NZ Office: Auckland, New Zealand
Telephone: 0800 800 806
Registration number
140 mg capsule: AUST R 228499
140 mg tablet: AUST R 319356
280 mg tablet: AUST R 319357
420 mg tablet: AUST R 319360
560 mg tablet: AUST R 319380
This leaflet was prepared in June 2022.
® IMBRUVICA is a registered trademark of Janssen-Cilag.
Co-developed with Pharmacyclics.
Published by MIMS July 2022


 After discontinuation of a CYP3A inhibitor, resume previous dose of Imbruvica (see Section 4.2 Dose and Method of Administration).
After discontinuation of a CYP3A inhibitor, resume previous dose of Imbruvica (see Section 4.2 Dose and Method of Administration).
 The efficacy data was further evaluated by an Independent Review Committee (IRC) demonstrating an ORR of 69%, with a 21% CR rate and a 48% PR rate. The IRC estimated median DOR was 19.6 months.
The efficacy data was further evaluated by an Independent Review Committee (IRC) demonstrating an ORR of 69%, with a 21% CR rate and a 48% PR rate. The IRC estimated median DOR was 19.6 months.
 A smaller proportion of patients treated with ibrutinib experienced a clinically meaningful worsening of lymphoma symptoms versus temsirolimus (27% versus 52%) and time to worsening of symptoms occurred more slowly with ibrutinib versus temsirolimus (HR 0.27, p < 0.0001).
A smaller proportion of patients treated with ibrutinib experienced a clinically meaningful worsening of lymphoma symptoms versus temsirolimus (27% versus 52%) and time to worsening of symptoms occurred more slowly with ibrutinib versus temsirolimus (HR 0.27, p < 0.0001).





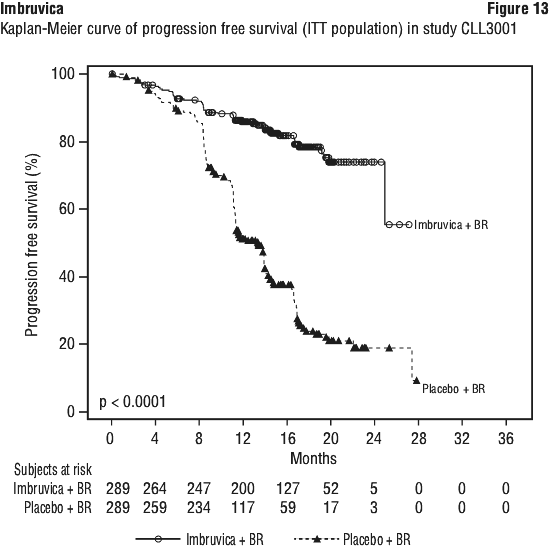
 The treatment effect of ibrutinib was consistent across the high-risk CLL/SLL population (del17p/TP53 mutation, del11q, or unmutated IGHV), with a PFS HR of 0.15 [95% CI (0.09, 0.27)], as shown in Table 9. The 2-year PFS rate estimates for the high-risk CLL/SLL population were 78.8% [95% CI (67.3, 86.7)] and 15.5% [95% CI (8.1, 25.2)] in the Imbruvica + obinutuzumab and chlorambucil + obinutuzumab arms, respectively.
The treatment effect of ibrutinib was consistent across the high-risk CLL/SLL population (del17p/TP53 mutation, del11q, or unmutated IGHV), with a PFS HR of 0.15 [95% CI (0.09, 0.27)], as shown in Table 9. The 2-year PFS rate estimates for the high-risk CLL/SLL population were 78.8% [95% CI (67.3, 86.7)] and 15.5% [95% CI (8.1, 25.2)] in the Imbruvica + obinutuzumab and chlorambucil + obinutuzumab arms, respectively. Any grade infusion-related reactions were observed in 25% of patients treated with Imbruvica + obinutuzumab and 58% of patients treated with chlorambucil + obinutuzumab. Grade 3 or higher or serious infusion-related reactions were observed in 3% of patients treated with Imbruvica + obinutuzumab and 9% of patients treated with chlorambucil + obinutuzumab.
Any grade infusion-related reactions were observed in 25% of patients treated with Imbruvica + obinutuzumab and 58% of patients treated with chlorambucil + obinutuzumab. Grade 3 or higher or serious infusion-related reactions were observed in 3% of patients treated with Imbruvica + obinutuzumab and 9% of patients treated with chlorambucil + obinutuzumab.
 With a median follow-up time on study of 49 months, median overall survival was not reached with a total of 23 deaths: 11 (3%) in the Imbruvica plus rituximab and 12 (7%) in the FCR treatment arms.
With a median follow-up time on study of 49 months, median overall survival was not reached with a total of 23 deaths: 11 (3%) in the Imbruvica plus rituximab and 12 (7%) in the FCR treatment arms.
 Across the high-risk CLL/SLL population (n=123), including TP53 mutation (n=9), del11q (n=38), or unmutated IGHV (n=109), the treatment effect of Imbruvica plus venetoclax was consistent, with a PFS HR of 0.23 [95% CI (0.13, 0.41)].
Across the high-risk CLL/SLL population (n=123), including TP53 mutation (n=9), del11q (n=38), or unmutated IGHV (n=109), the treatment effect of Imbruvica plus venetoclax was consistent, with a PFS HR of 0.23 [95% CI (0.13, 0.41)]. Twelve months after the completion of treatment, MRD negativity rates in peripheral blood were 49.1% (52/106) by NGS assay and 54.7% (58/106) by flow cytometry in patients treated with Imbruvica plus venetoclax and, at the corresponding time point, was 12.4% (13/105) by NGS assay and 16.2% (17/105) by flow cytometry in patients treated with chlorambucil plus obinutuzumab.
Twelve months after the completion of treatment, MRD negativity rates in peripheral blood were 49.1% (52/106) by NGS assay and 54.7% (58/106) by flow cytometry in patients treated with Imbruvica plus venetoclax and, at the corresponding time point, was 12.4% (13/105) by NGS assay and 16.2% (17/105) by flow cytometry in patients treated with chlorambucil plus obinutuzumab. The efficacy data were further evaluated using IWCLL criteria by an independent review committee (IRC), demonstrating an ORR of 64.7% (95% CI: 50.1%, 77.6%), all partial responses. The DOR ranged from 3.9 to 24.2+ months. The median DOR was not reached.
The efficacy data were further evaluated using IWCLL criteria by an independent review committee (IRC), demonstrating an ORR of 64.7% (95% CI: 50.1%, 77.6%), all partial responses. The DOR ranged from 3.9 to 24.2+ months. The median DOR was not reached. The Kaplan-Meier curves for PFS and OS are shown in Figures 9 and 10, respectively.
The Kaplan-Meier curves for PFS and OS are shown in Figures 9 and 10, respectively.
 The efficacy was similar across all of the subgroups examined, including in patients with and without deletion 17p, a pre-specified stratification factor (Figure 11).
The efficacy was similar across all of the subgroups examined, including in patients with and without deletion 17p, a pre-specified stratification factor (Figure 11).


 The median time to response was 1.0 month (range: 0.7-13.4 months).
The median time to response was 1.0 month (range: 0.7-13.4 months).
 Tumor flare in the form of IgM increase occurred in 8.0% of subjects in the Imbruvica + rituximab arm and 46.7% of subjects in the placebo + rituximab arm.
Tumor flare in the form of IgM increase occurred in 8.0% of subjects in the Imbruvica + rituximab arm and 46.7% of subjects in the placebo + rituximab arm. The chemical name of the ibrutinib is 1 [(3R)-3-[4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3, 4-d]pyrimidin-1-yl]- 1-piperidinyl]-2-propen-1-one.
The chemical name of the ibrutinib is 1 [(3R)-3-[4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3, 4-d]pyrimidin-1-yl]- 1-piperidinyl]-2-propen-1-one.