What is in this leaflet
This leaflet answers some common questions people ask about IMFINZI. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor will have weighed the risks of you taking IMFINZI against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor.
Keep this leaflet, you may need to read it again.
What IMFINZI is used for
This medicine is used to treat a type of lung cancer called Non-Small Cell Lung Cancer. It will be prescribed to you if:
- your cancer has spread within your lung and cannot be removed by surgery and;
- you have tried radiation and chemotherapy that contains platinum, and your cancer has shrunk or has not worsened.
This medicine is also used to treat a type of lung cancer called Extensive-Stage Small Cell Lung Cancer. It will be prescribed to you if your cancer has spread within
- your lungs (or to other parts of the body) and
- you have not received previous treatment.
IMFINZI will be given in combination with chemotherapy for this indication. It is important that you also read the consumer leaflets for the specific chemotherapy you will be receiving. If you have any questions about these medicines, ask your doctor.
IMFINZI is a monoclonal antibody, a type of protein. It is a type of immunotherapy and belongs to a group of medicines called immune checkpoint inhibitors. It works with your immune system to destroy cancer cells.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
This medicine is not expected to affect your ability to drive a car or operate machinery.
Before you are given IMFINZI
When you must not be given this medicine
You should not be given this medicine if you have an allergy to:
- durvalumab or any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
You should not be given this medicine if you are pregnant. It may affect your developing baby if you use it during pregnancy.
You should not be given this medicine if you are breast-feeding. It is not known if the active ingredient in IMFINZI passes into breast milk, but if it does, there is a possibility that your baby may be affected.
If you are not sure whether you should be given this medicine, talk to your doctor.
Before you are given this medicine
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you:
- have immune system problems such as Crohn's disease, ulcerative colitis, or lupus
- have had an organ transplant
- have lung or breathing problems
- have liver problems.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding or plan to breast-feed. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell them before you are given IMFINZI.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
How IMFINZI is given to you
IMFINZI will be given to you as a liquid infusion into your vein (IV). An infusion takes about 1 hour and will normally be given every 2, 3 or 4 weeks.
Your doctor will decide how many treatments you need.
If you miss an appointment to be given IMFINZI
Call your doctor right away to reschedule your appointment.
It is very important that you do not miss a dose of this medicine.
While you are being treated IMFINZI
Things you must do
Tell any other doctors, dentists, and pharmacists who treat you that you are using this medicine.
If you become pregnant while using this medicine, tell your doctor immediately.
If you are a woman who could become pregnant, you must use adequate birth control while you are being treated with this medicine and for at least 3 months after your last dose.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not breast-feed if you are being given this medicine and for at least 3 months after the last dose. It is not known if the active ingredient in IMFINZI passes into breast milk, but if it does, there is a possibility that your baby may be affected.
Side effects
This medicine may have unwanted side effects in some people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
IMFINZI can cause your immune system to attack normal organs and tissues in many areas of your body and can affect the way they work and this can cause the side effects.
If you have any of the following symptoms, call or see your doctor right away. Your doctor may give you other medicines in order to prevent more severe complications and reduce your symptoms. Your doctor may withhold the next dose of IMFINZI or stop your treatment with IMFINZI.
- new or worsening cough, shortness of breath, or chest pain which may indicate inflammation of your lungs
- nausea or vomiting, feeling less hungry, pain on the right side of stomach, yellowing of skin or whites of eyes, drowsiness, dark urine, or bleeding or bruising more easily than normal which may indicate inflammation of your liver
- diarrhoea or more bowel movements than usual, black, tarry, sticky stools or stools with blood or mucus, severe stomach pain or tenderness which may indicate inflammation of your intestines
- headaches that will not go away or unusual headaches, extreme tiredness, weight gain or weight loss, dizziness or fainting, feeling more hungry or thirsty than usual, hair loss, feeling cold, constipation, changes in your voice, urinating more often than usual, nausea or vomiting, stomach area (abdomen) pain, changes in mood or behaviour, such as decreased sex drive, irritability, or forgetfulness, fast and deep breathing, confusion, or a sweet smell to your breath, a sweet or metallic taste in your mouth or a different odour to your urine or sweat. These may indicate inflammation of some of your hormone glands
- changes in the amount or colour of your urine, swelling in your ankles, or loss of appetite may indicate inflammation of your kidneys
- the need to urinate urgently and frequently, burning pain or sensation when urinating, bladder still feels full after urinating, pain above your pubic bone, blood in the urine, all of which may indicate a urinary tract infection
- rash, itching, skin blistering, or ulcers in mouth or other mucous membranes which may indicate inflammation of the skin or mouth
- chest pain, shortness of breath, or irregular heartbeat which may indicate inflammation of the heart
- chills or shaking, itching or rash, flushing, shortness of breath or wheezing dizziness, fever, feeling like passing out, back or neck pain, or facial swelling which may indicate infusion related reactions
- muscle weakness, tiredness and/or pain, and/or rapid fatigue of the muscles, in one or more areas of your body which may indicate inflammation or problems of the muscles
- bleeding (nose or gum bleeding) and/or bruising may indicate low number of platelets
- seizures, neck stiffness, headache, fever, chills, vomiting, eye sensitivity to light, confusion and sleepiness which may indicate encephalitis or meningitis
- pain, weakness, and paralysis in the extremities which may indicate Guillain-Barré syndrome.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
The most frequent serious side-effects experienced when IMFINZI was given alone are:
- muscle and bone pain
- problems with your liver
- general physical health deterioration
- infections
- pain in your stomach area
- too much calcium in your blood
- vomiting
- serious lung infections (pneumonia)
- inflammation of the lung (pneumonitis)
- fever.
The most common side-effects experienced when IMFINZI was given alone are:
- muscle and bone pain
- problems with your liver
- upper respiratory tract infection such as in the nose or throat e.g. sinusitis or tonsilitis
- lung infections which may be serious such as pneumonia or influenza
- thrush in the mouth
- infusion related reactions
- feeling tired
- constipation
- decreased appetite
- nausea
- not enough red blood cells (anaemia)
- diarrhoea and colitis (bowel inflammation)
- rash or itchiness
- swellings, especially on your legs or hands
- shortness of breath
- cough
- fever
- not enough sodium in your blood
- insomnia
- underactive thyroid gland that can cause tiredness or weight gain
- overactive thyroid gland that can cause rapid heartbeat, weight loss, increased appetite and anxiety
- pain in your stomach area.
The most frequent serious side-effects experienced when IMFINZI was given in combination with chemotherapy are:
- not enough white blood cells with fever (febrile neutropenia)
- serious lung infections (pneumonia)
- not enough red blood cells (anaemia)
- not enough red blood cells, white blood cells, and platelets (pancytopenia)
- inflammation of the lung (pneumonitis)
- Chronic Obstructive Pulmonary Disorder (COPD) which causes the narrowing of airways and makes it difficult to breathe.
The most common side-effects experienced when IMFINZI was given in combination with chemotherapy are:
- not enough white blood cells (neutropenia and leukopenia)
- not enough red blood cells (anaemia)
- nausea
- hair loss
- decreased appetite
- feeling tired or weak
- constipation
- not enough platelets (thrombocytopenia)
- vomiting
- cough.
Ask your doctor to answer any questions you may have about side effects.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some people.
Some side effects can only be found when your doctor does blood tests from time to time to check your progress (for example, low white blood cell count, low red blood cell count and low platelet count).
After being given IMFINZI
Do not breast feed for at least 3 months after the last dose.
If you are a woman who could become pregnant, you must use adequate birth control for at least 3 months after your last dose.
Product description
What it looks like
IMFINZI concentrated solution for infusion is a clear to opalescent, colourless to slightly yellow liquid in a glass vial.
Ingredients
IMFINZI contains durvalumab as the active ingredient.
Other ingredients include:
- histidine
- histidine hydrochloride monohydrate
- trehalose dihydrate
- polysorbate 80
- water for injection.
How to store IMFINZI
Store unopened vials under refrigeration at 2°C to 8°C in the original carton to protect from light. Do not freeze. Do not shake.
Supplier
IMFINZI is sponsored and supplied in Australia by:
AstraZeneca Pty Ltd
ABN 54 009 682 311
66 Talavera Road
MACQUARIE PARK NSW 2113
Telephone: 1800 805 342
This leaflet was prepared on
September 2021
Australian Registration Number(s):
IMFINZI, 500 mg (500 mg/10mL) in 10 mL vial for intravenous infusion - AUST R 283216
IMFINZI, 120 mg (120 mg/2.4mL) in 10 mL vial for intravenous infusion - AUST R 283215
© AstraZeneca 2021
Doc ID-003924994 v12
Published by MIMS November 2021
 It is recommended to continue treatment for clinically stable patients with initial evidence of disease progression until disease progression is confirmed.
It is recommended to continue treatment for clinically stable patients with initial evidence of disease progression until disease progression is confirmed. After withhold, Imfinzi can be resumed within 12 weeks if the ADR improved to ≤ Grade 1 and the corticosteroid dose has been reduced to ≤ 10 mg prednisone or equivalent per day. Imfinzi should be permanently discontinued for recurrent Grade 3 ADR, as applicable.
After withhold, Imfinzi can be resumed within 12 weeks if the ADR improved to ≤ Grade 1 and the corticosteroid dose has been reduced to ≤ 10 mg prednisone or equivalent per day. Imfinzi should be permanently discontinued for recurrent Grade 3 ADR, as applicable.
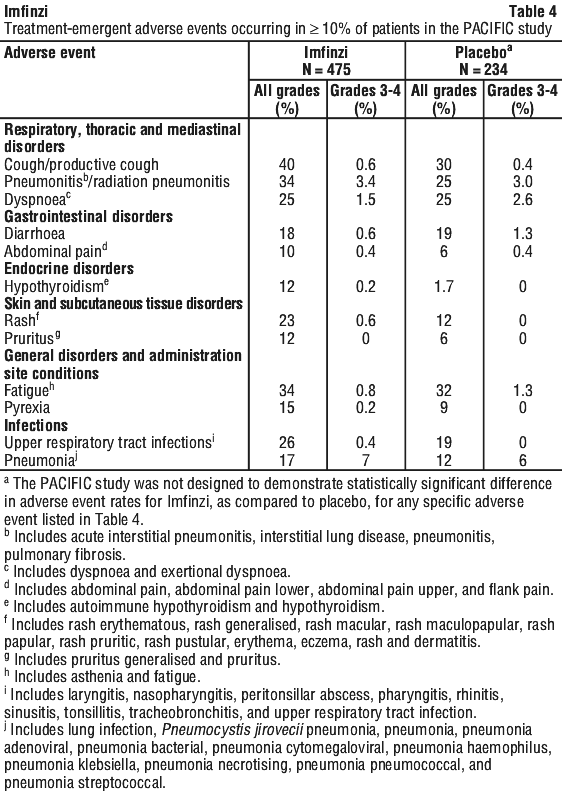 Other adverse events occurring in less than 10% of patients treated with Imfinzi were dysphonia, dysuria, night sweats, peripheral oedema, and increased susceptibility to infections.
Other adverse events occurring in less than 10% of patients treated with Imfinzi were dysphonia, dysuria, night sweats, peripheral oedema, and increased susceptibility to infections.



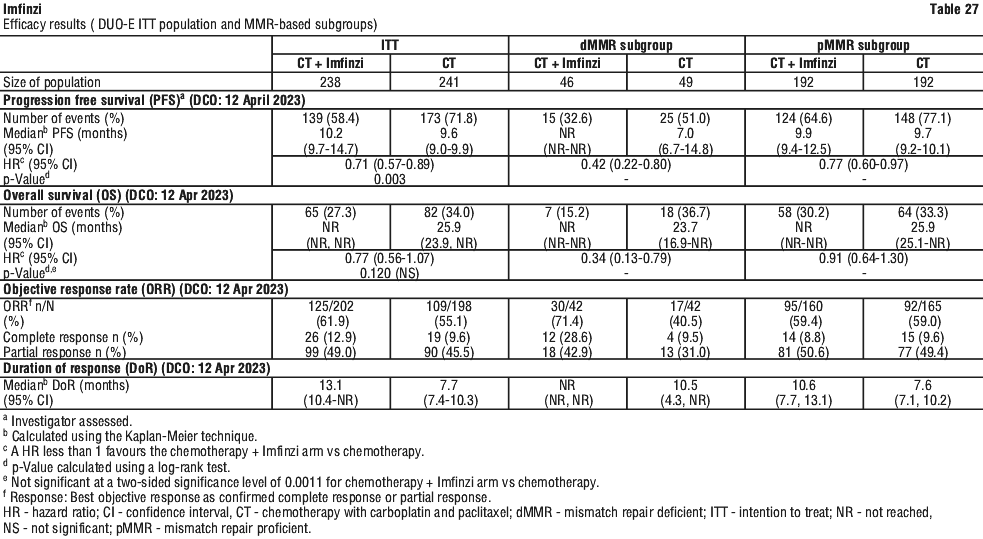
 Table 10 summarises the laboratory abnormalities that occurred in at least 20% of patients treated with Imfinzi plus chemotherapy.
Table 10 summarises the laboratory abnormalities that occurred in at least 20% of patients treated with Imfinzi plus chemotherapy. The proportion of patients who experienced a TSH shift from baseline that was ≤ ULN to any grade > ULN was 17.7% and a TSH shift from baseline that was ≥ LLN to any grade < LLN was 31.3%.
The proportion of patients who experienced a TSH shift from baseline that was ≤ ULN to any grade > ULN was 17.7% and a TSH shift from baseline that was ≥ LLN to any grade < LLN was 31.3%. Table 12 summarises the laboratory abnormalities that occurred in at least 20% of patients treated with Imfinzi plus chemotherapy.
Table 12 summarises the laboratory abnormalities that occurred in at least 20% of patients treated with Imfinzi plus chemotherapy.
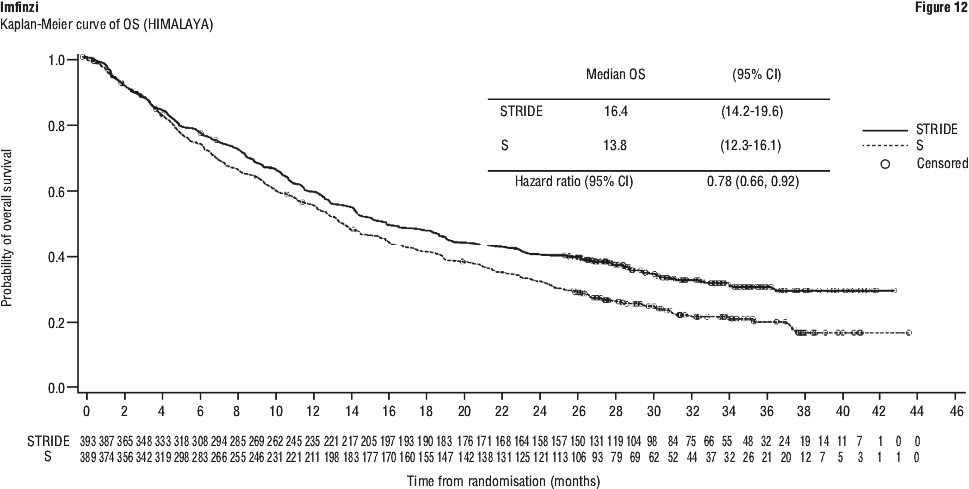
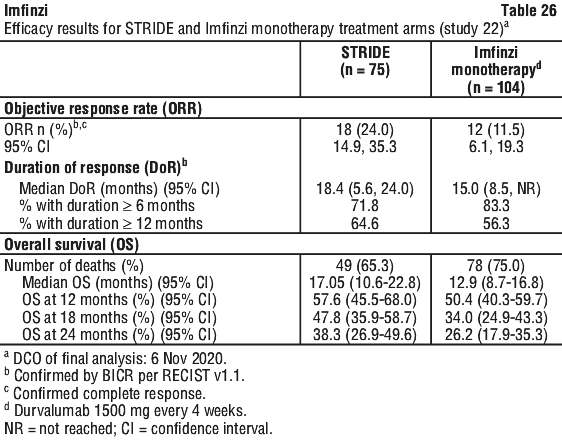
 Table 14 summarises the laboratory abnormalities that occurred patients treated with STRIDE in the HIMALAYA study.
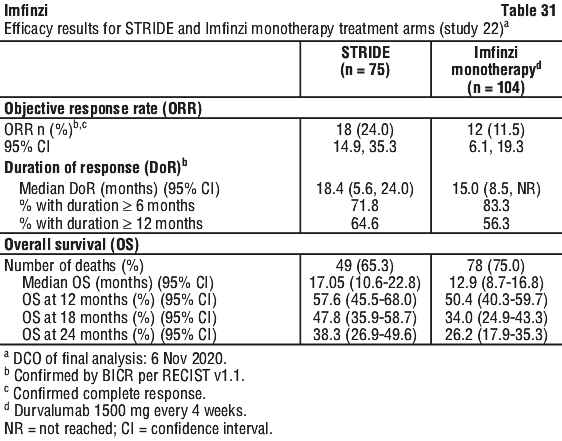
Table 14 summarises the laboratory abnormalities that occurred patients treated with STRIDE in the HIMALAYA study. Table 15 summarises the adverse events that occurred in patients treated with STRIDE in Study 22.
Table 15 summarises the adverse events that occurred in patients treated with STRIDE in Study 22. Table 16 summarises the laboratory abnormalities that occurred patients treated with STRIDE in Study 22.
Table 16 summarises the laboratory abnormalities that occurred patients treated with STRIDE in Study 22.





 Serious adverse drug reactions (≥ 2%) in the Imfinzi arm were diarrhoea (2.5%) during the neoadjuvant phase, and pneumonia (2.5%) in the adjuvant phase.
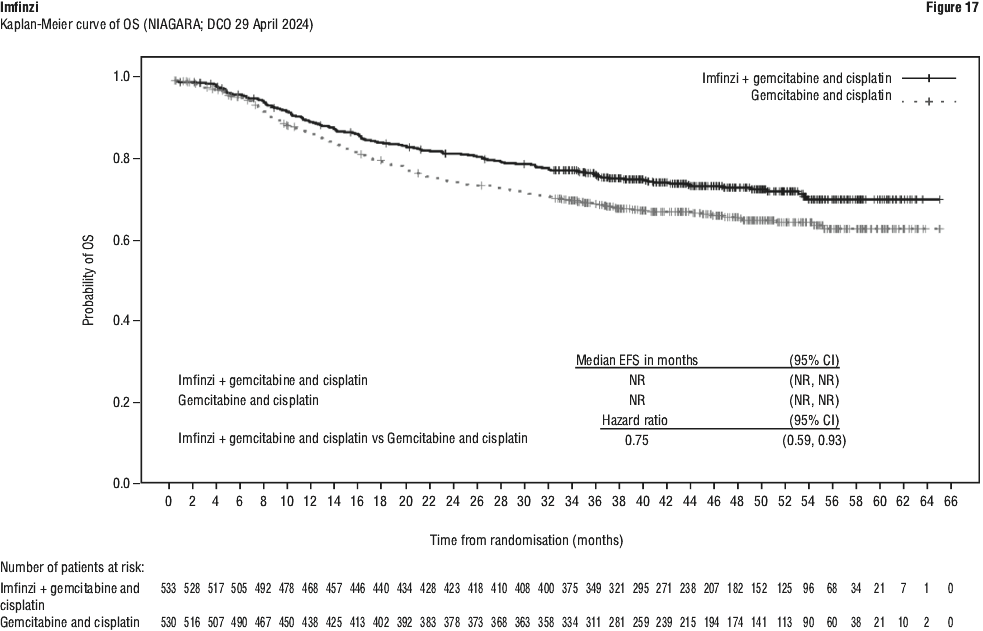
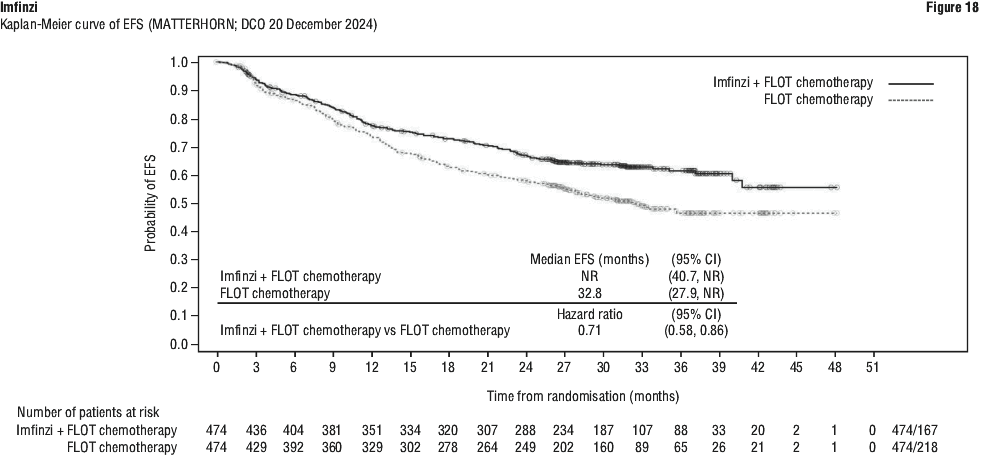
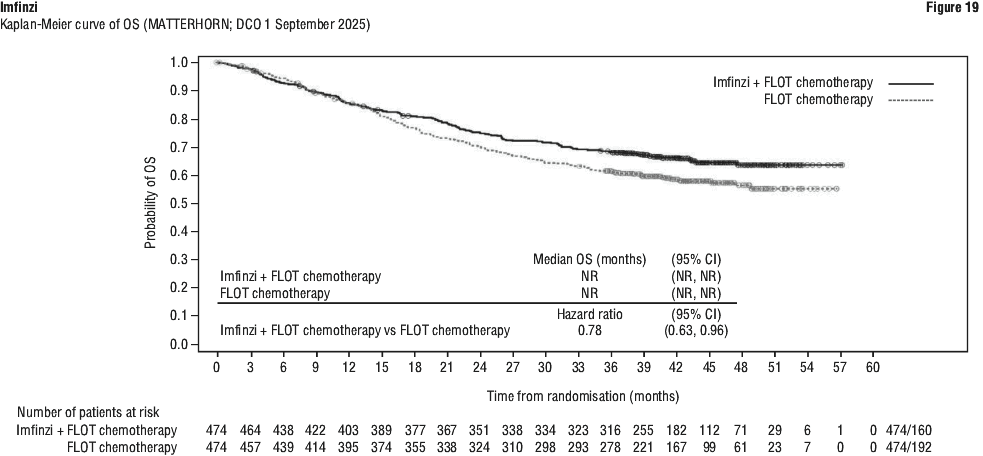
Serious adverse drug reactions (≥ 2%) in the Imfinzi arm were diarrhoea (2.5%) during the neoadjuvant phase, and pneumonia (2.5%) in the adjuvant phase.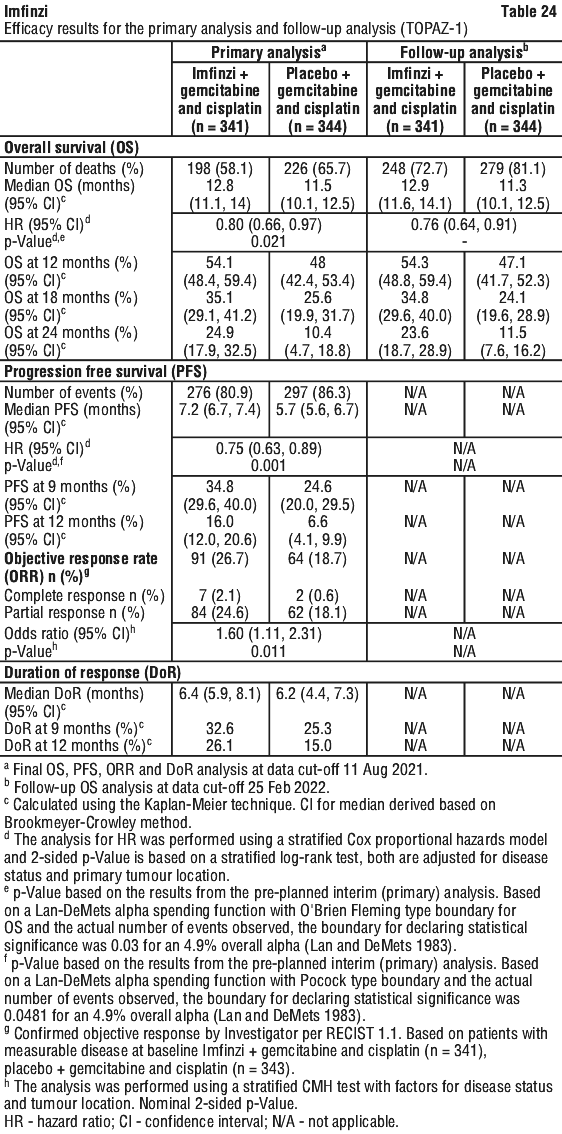 The study demonstrated a statistically significant and clinically meaningful improvement in EFS of the Imfinzi arm compared to the placebo arm. The study also demonstrated a statistically significant and meaningful improvement in pCR of the Imfinzi arm compared to the placebo arm. OS data were not mature at the time of EFS analysis. See Table 25 and Figure 1.
The study demonstrated a statistically significant and clinically meaningful improvement in EFS of the Imfinzi arm compared to the placebo arm. The study also demonstrated a statistically significant and meaningful improvement in pCR of the Imfinzi arm compared to the placebo arm. OS data were not mature at the time of EFS analysis. See Table 25 and Figure 1.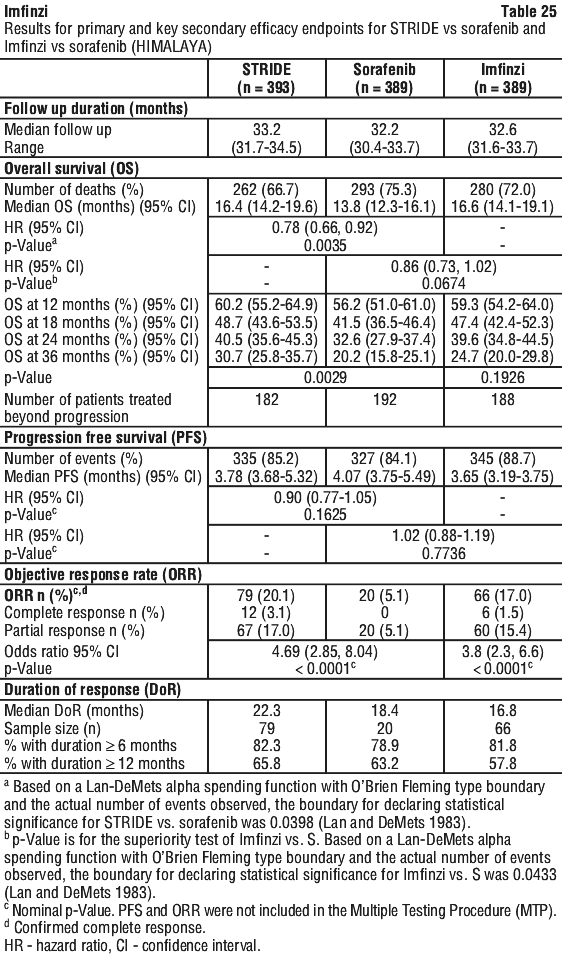


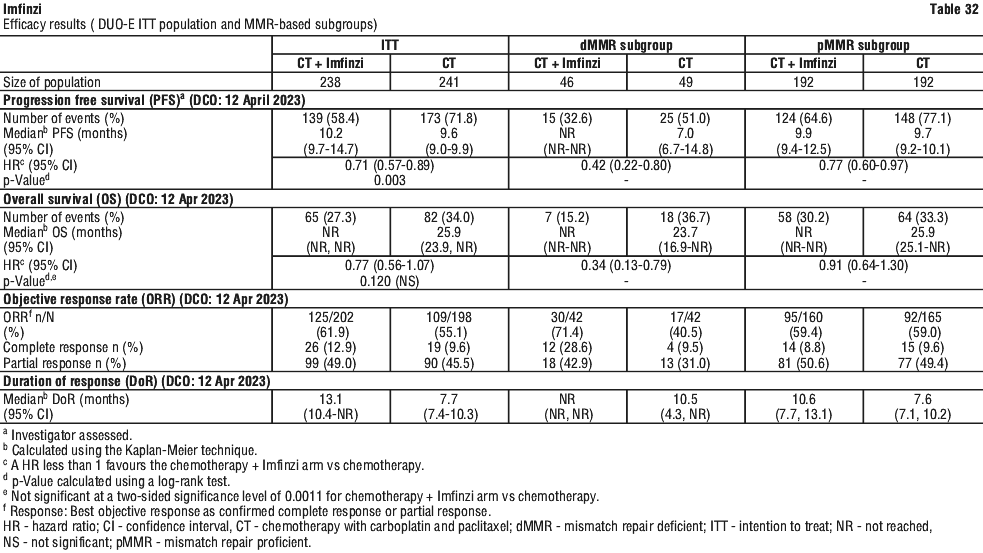
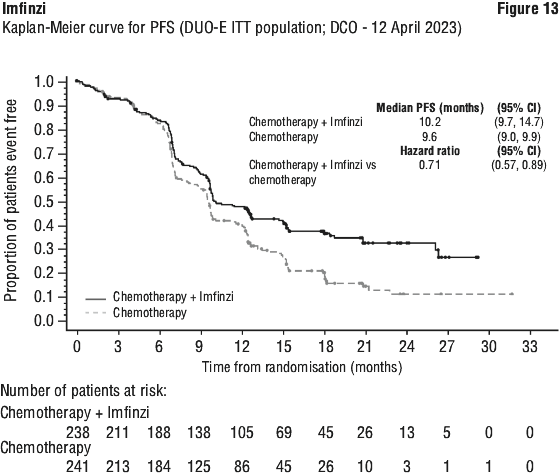
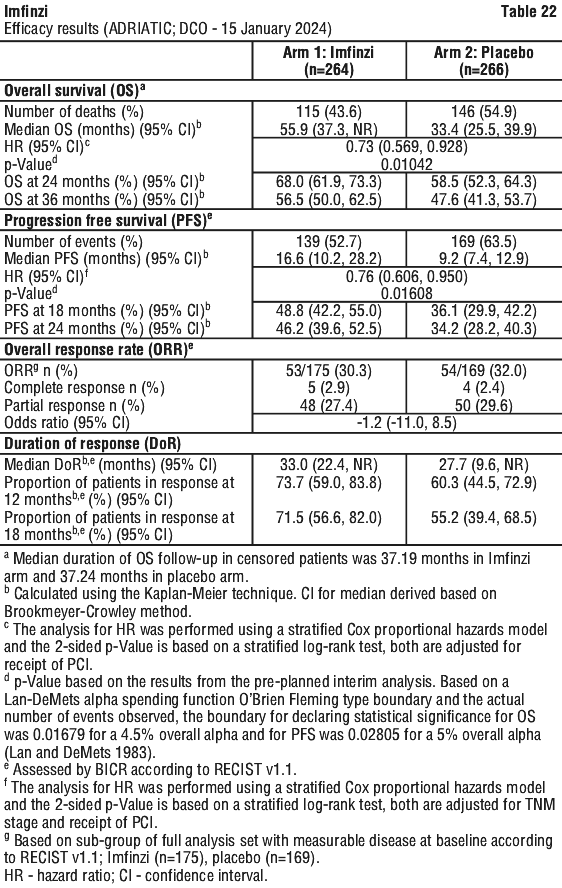
 The improvements in PFS and OS in favour of patients receiving Imfinzi compared to those receiving placebo were consistently observed in all predefined subgroups analysed, including ethnicity, age, gender, smoking history, EGFR mutation status and histology. ALK mutation status was not analysed in this study.
The improvements in PFS and OS in favour of patients receiving Imfinzi compared to those receiving placebo were consistently observed in all predefined subgroups analysed, including ethnicity, age, gender, smoking history, EGFR mutation status and histology. ALK mutation status was not analysed in this study.


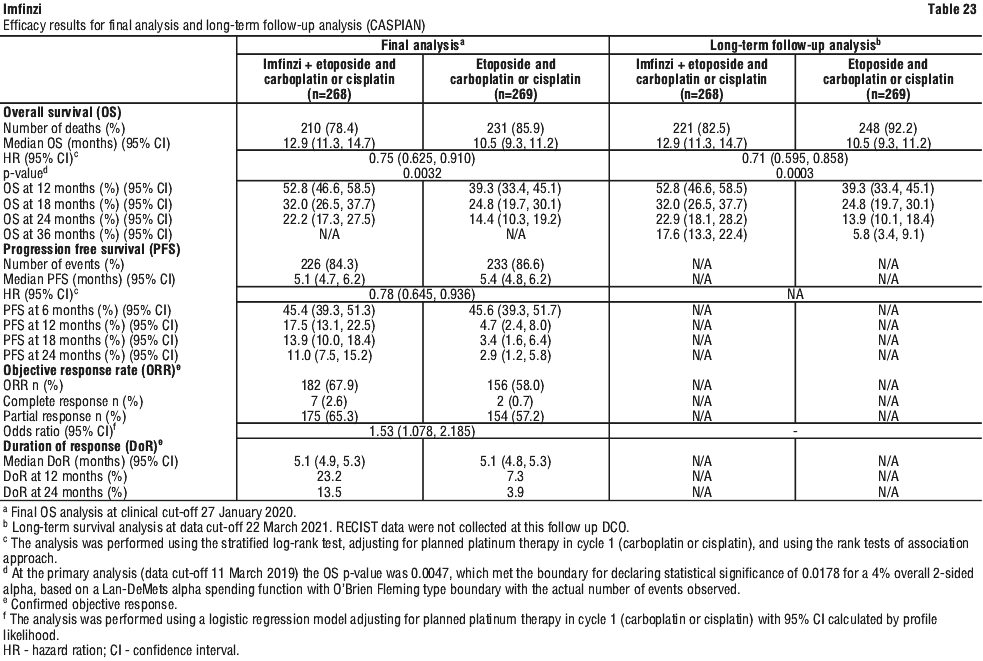
 The improvements in OS and PFS in favour of patients receiving Imfinzi compared to those receiving placebo were generally consistent across predefined subgroups analysed.
The improvements in OS and PFS in favour of patients receiving Imfinzi compared to those receiving placebo were generally consistent across predefined subgroups analysed.