1 Name of Medicine
Tremelimumab.
2 Qualitative and Quantitative Composition
Each mL contains 20 mg of Imjudo.
Each vial of 1.25 mL contains 25 mg of tremelimumab.
Each vial of 15 mL contains 300 mg of tremelimumab.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Injection, concentrated.
Sterile, preservative-free, clear to slightly opalescent, colourless to slightly yellow solution, free from or practically free from visible particles.
4.1 Therapeutic Indications
Hepatocellular carcinoma (HCC).
Imjudo in combination with durvalumab is indicated for the treatment of adult patients with unresectable hepatocellular carcinoma (uHCC) who have not received prior treatment with a PD-1/PD-L1 inhibitor.4.2 Dose and Method of Administration
The recommended dose of Imjudo is presented in Table 1.
Imjudo is administered as an intravenous infusion over 1 hour.
Imjudo is used in combination with durvalumab. Refer to the Product Information for durvalumab for its recommended dosing information.
The proposed combination should be administered and monitored under the supervision of physicians experienced with the use of immunotherapy.
 Dose reduction or escalation is not recommended during treatment with Imjudo in combination with durvalumab. Treatment withholding or discontinuation may be required based on individual safety and tolerability.
Dose reduction or escalation is not recommended during treatment with Imjudo in combination with durvalumab. Treatment withholding or discontinuation may be required based on individual safety and tolerability.
Immune-mediated adverse reactions requiring specific treatment modification and management are summarised in Table 2. See Section 4.4 Special Warnings and Precautions for Use for further management recommendations, monitoring and evaluation information.
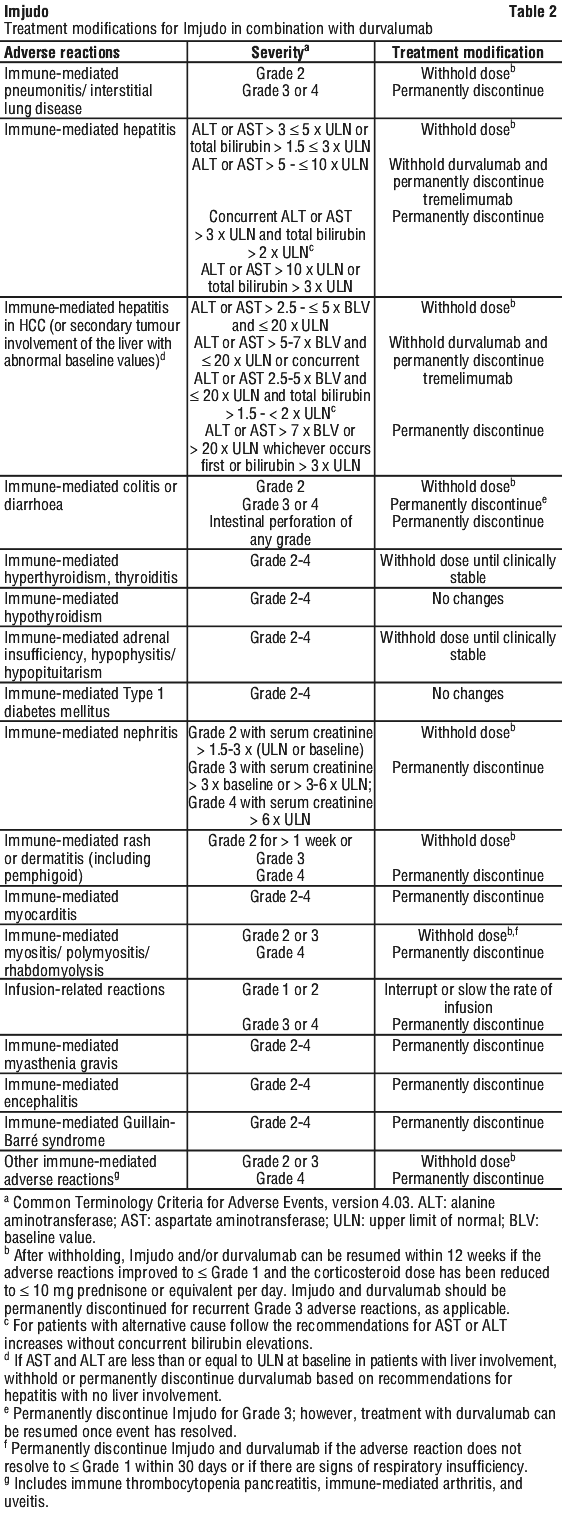 For non-immune-mediated adverse reactions, withhold Imjudo and/or durvalumab for Grade 2 and 3 adverse reactions until ≤ Grade 1 or return to baseline. Imjudo and durvalumab should be discontinued for Grade 4 adverse reactions (with the exception of Grade 4 laboratory abnormalities, about which the decision to discontinue should be based on accompanying clinical signs/symptoms and clinical judgment.
For non-immune-mediated adverse reactions, withhold Imjudo and/or durvalumab for Grade 2 and 3 adverse reactions until ≤ Grade 1 or return to baseline. Imjudo and durvalumab should be discontinued for Grade 4 adverse reactions (with the exception of Grade 4 laboratory abnormalities, about which the decision to discontinue should be based on accompanying clinical signs/symptoms and clinical judgment.
Special patient populations.
Based on a population pharmacokinetic analysis, no dose adjustment of Imjudo is recommended based on patient age, body weight, gender and race (see Section 5.2 Pharmacokinetic Properties).
Renal impairment.
Based on a population pharmacokinetic analysis, no dose adjustment of Imjudo is recommended in patients with mild or moderate renal impairment. Imjudo has not been studied in patients with severe renal impairment (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
Based on a population pharmacokinetic analysis, no dose adjustment of Imjudo is recommended for patients with mild or moderate hepatic impairment. Imjudo has not been studied in patients with severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Use in the elderly.
No dose adjustment is required for elderly patients (≥ 65 years of age) (see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties).
Paediatric use.
The safety and effectiveness of Imjudo have not been established in patients less than 18 years.
Method of administration.
Preparation of solution.
Imjudo is supplied as a single-dose vial and does not contain any preservatives, aseptic technique must be observed.
Visually inspect drug product for particulate matter and discolouration. Imjudo is clear to slightly opalescent, colourless to slightly yellow solution. Discard the vial if the solution is cloudy, discoloured or visible particles are observed. Do not shake the vial.
Withdraw the required volume from the vial(s) of Imjudo and transfer into an intravenous (IV) bag containing 0.9% sodium chloride Injection, or 5% dextrose injection. Mix diluted solution by gentle inversion. The final concentration of the diluted solution should be between 0.1 mg/mL and 10 mg/mL. Do not freeze or shake the solution.
Care must be taken to ensure the sterility of prepared solutions.
Do not re-enter the vial after withdrawal of drug.
Discard any unused portion left in the vial.
After preparation of infusion solution.
Imjudo does not contain a preservative. Administer infusion solution immediately once prepared. If infusion solution is not administered immediately and it needs to be stored, the total time from preparation to the start of administration should not exceed:
28 days in a refrigerator at 2°C to 8°C.
24 hours at room temperature up to 30°C.
Administration.
Imjudo is for single use in one patient only. Discard any residue.
Administer infusion solution intravenously over 1 hour through an intravenous line containing a sterile, low-protein binding 0.2 or 0.22 micron filter.
Do not co-administer other drugs through the same infusion line.
When Imjudo is administered in combination with durvalumab, administer Imjudo prior to durvalumab on the same day. Imjudo and durvalumab are administered as separate intravenous infusions.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Immune-mediated adverse reactions.
See Section 4.2 Dose and Method of Administration, Table 2 for recommended treatment modifications.
For suspected immune-mediated adverse reactions, adequate evaluation should be performed to confirm aetiology or exclude alternate aetiologies. Based on the severity of the adverse reaction, Imjudo or Imjudo in combination with durvalumab should be withheld and corticosteroids administered. Upon improvement to ≤ Grade 1, corticosteroid taper should be initiated and continued over at least 1 month. Consider increasing dose of corticosteroids and/or using additional systemic immunosuppressants if there is worsening or no improvement.
The Imjudo Consumer Medicine Information (CMI) should be provided to the patient prior to administration of the first dose of Imjudo.
Immune-mediated pneumonitis. Immune-mediated pneumonitis or interstitial lung disease, defined as requiring use of systemic corticosteroids and with no clear alternate aetiology, occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of pneumonitis. Suspected pneumonitis should be confirmed with radiographic imaging and other infectious and disease-related aetiologies excluded, and managed as recommended in Section 4.2 Dose and Method of Administration. For Grade 2 events, an initial dose of 1-2 mg/kg/day prednisone or equivalent should be initiated followed by a taper. For Grade 3 or 4 events, an initial dose of 2-4 mg/kg/day methylprednisolone or equivalent (or in accordance with local immune-related adverse events management guidelines where these differ) should be initiated followed by a taper.
Immune-mediated hepatitis. Immune-mediated hepatitis, defined as requiring use of systemic corticosteroids and with no clear alternate aetiology, occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for abnormal liver tests prior to and periodically during treatment with Imjudo in combination with durvalumab. Immune-mediated hepatitis should be managed as recommended in Section 4.2 Dose and Method of Administration. Corticosteroids should be administered with an initial dose of 1-2 mg/kg/day prednisone or equivalent followed by taper for all grades.
Immune-mediated colitis. Immune-mediated colitis or diarrhoea, defined as requiring use of systemic corticosteroids and with no clear alternate aetiology, occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Intestinal perforation and large intestine perforation were reported in patients receiving Imjudo in combination with durvalumab. Patients should be monitored for signs and symptoms of colitis/diarrhoea and intestinal perforation and managed as recommended in Section 4.2 Dose and Method of Administration. Corticosteroids should be administered at an initial dose of 1-2 mg/kg/day prednisone or equivalent followed by a taper for Grades 2-4. Consult a surgeon immediately if intestinal perforation of any grade is suspected.
Immune-mediated endocrinopathies. Immune-mediated hypothyroidism/ hyperthyroidism/ thyroiditis.
Immune-mediated hypothyroidism, hyperthyroidism or thyroiditis have occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for abnormal thyroid function tests prior to and periodically during treatment and managed as recommended in Section 4.2 Dose and Method of Administration. For immune-mediated hypothyroidism, initiate thyroid hormone replacement as clinically indicated for Grades 2-4. For immune-mediated hyperthyroidism/thyroiditis, symptomatic management can be implemented for Grades 2-4.
Immune-mediated adrenal insufficiency.
Immune-mediated adrenal insufficiency occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for clinical signs and symptoms of adrenal insufficiency and managed as recommended in Section 4.2 Dose and Method of Administration. Corticosteroids should be administered for with an initial dose of 1-2 mg/kg/day prednisone or equivalent followed by taper and a hormone replacement as clinically indicated for Grades 2-4.
Immune-mediated type 1 diabetes mellitus.
Immune-mediated type 1 diabetes mellitus, which can present with diabetic ketoacidosis, occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for clinical signs and symptoms of type 1 diabetes mellitus and managed as recommended in Section 4.2 Dose and Method of Administration. Treatment with insulin can be initiated as clinically indicated for Grades 2-4.
Immune-mediated hypophysitis/ hypopituitarism.
Immune-mediated hypophysitis or hypopituitarism occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for clinical signs and symptoms of hypophysitis or hypopituitarism and managed as recommended in Section 4.2 Dose and Method of Administration. Corticosteroids should be administered for with an initial dose of 1-2 mg/kg/day prednisone or equivalent followed by taper and a hormone replacement as clinically indicated for Grades 2-4.
Immune-mediated nephritis. Immune-mediated nephritis, defined as requiring use of systemic corticosteroids and with no clear alternate aetiology, occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for abnormal renal function tests prior to and periodically during treatment with Imjudo in combination with durvalumab and managed as recommended in Section 4.2 Dose and Method of Administration. Corticosteroids should be administered with an initial dose of 1-2 mg/kg/day prednisone or equivalent followed by taper for Grades 2-4.
Immune-mediated rash. Immune-mediated rash or dermatitis (including pemphigoid), defined as requiring use of systemic corticosteroids and with no clear alternate aetiology, occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of rash or dermatitis and managed as recommended in Section 4.2 Dose and Method of Administration. Corticosteroids should be administered with an initial dose of 1-2 mg/kg/day prednisone or equivalent followed by taper for Grade 2 > 1 week or Grade 3 and 4.
Immune-mediated myocarditis. Immune-mediated myocarditis, which can be fatal, occurred in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of immune-mediated myocarditis and managed as recommended in Section 4.2 Dose and Method of Administration. Corticosteroids should be administered with an initial dose of 2-4 mg/kg/day prednisone or equivalent followed by taper for Grades 2-4. If no improvement within 2 to 3 days despite corticosteroids, promptly start additional immunosuppressive therapy. Upon resolution (Grade 0), corticosteroid taper should be initiated and continued over at least 1 month.
Other immune-mediated adverse reactions. Given the mechanism of action of Imjudo and durvalumab, other potential immune-mediated adverse reactions may occur in patients receiving the combination of Imjudo with durvalumab. The following immune-mediated adverse reactions have been observed: myasthenia gravis, myositis, polymyositis, rhabdomyolysis, Guillain-Barré syndrome, immune thrombocytopenia, pancreatitis, immune-mediated arthritis, uveitis and encephalitis (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms and managed as recommended in Section 4.2 Dose and Method of Administration. Corticosteroids should be administered an initial dose of 1-2 mg/kg/day prednisone or equivalent followed by taper for Grades 2-4.
Patients with pre-existing autoimmune disease (AID).
In patients with pre-existing autoimmune disease (AID), data from observational studies suggest that the risk of immune-mediated adverse reactions following immune checkpoint inhibitor therapy may be increased as compared with the risk in patients without pre-existing AID. In addition, flares of the underlying AID were frequent, but the majority were mild and manageable.
Infusion-related reactions.
Patients should be monitored for signs and symptoms of infusion-related reactions and managed as recommended in Section 4.2 Dose and Method of Administration. Severe infusion-related reactions have been reported in patients receiving Imjudo in combination with durvalumab (see Section 4.8 Adverse Effects (Undesirable Effects)). For Grade 1 or 2 severity, may consider pre-medications for prophylaxis of subsequent infusion reactions. For Grade 3 or 4, manage severe infusion-related reactions per institutional standard, appropriate clinical practice guidelines and/or society guidelines.
Use in the elderly.
No dose adjustment is required for elderly patients (≥ 65 years of age). Of the 462 patients with uHCC treated with STRIDE, 173 patients were 65 years or older. No overall clinically meaningful differences in safety or efficacy were reported between patients ≥ 65 years of age and younger patients.
Paediatric use.
The safety and effectiveness of Imjudo have not been established in patients aged less than 18 years.
Patients excluded from clinical studies.
Patients with the following were excluded from clinical studies: Child-Pugh Score B or C, main portal vein thrombosis, liver transplant, uncontrolled hypertension, history of, or current brain metastases, spinal cord compression, co-infection of viral hepatitis B and hepatitis C, active or prior documented gastrointestinal (GI) bleeding within 12 months, ascites requiring non-pharmacologic intervention within 6 months, hepatic encephalopathy within 12 months before the start of treatment, active or prior documented autoimmune or inflammatory disorders. In the absence of data, tremelimumab should be used with caution in these populations after careful consideration of the potential benefit/risk on an individual basis.
Sodium content.
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, that is to say essentially 'sodium-free'.
Effects on laboratory tests.
Please see Section 4.8 Adverse Effects (Undesirable Effects).4.5 Interactions with Other Medicines and Other Forms of Interactions
Tremelimumab is an immunoglobulin and the primary elimination pathways of tremelimumab are protein catabolism via reticuloendothelial system or target-mediated disposition; therefore, no formal pharmacokinetic (PK) drug-drug interaction studies have been conducted with tremelimumab since no metabolic drug-drug interactions are expected. PK drug-drug interaction between tremelimumab in combination with durvalumab was assessed in the HIMALAYA study and no clinically meaningful PK drug-drug interaction was identified.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no data on the potential effects of tremelimumab on fertility in humans.
Animal fertility studies have not been conducted with tremelimumab. Thus, the effect of tremelimumab on male and female fertility is unknown.
(Category D)
There are no data on the use of tremelimumab in pregnant women. Based on its mechanism of action and findings from animal studies with drugs of same pharmacological class, tremelimumab has the potential to impact pregnancy maintenance and may cause fetal harm when administered to a pregnant woman. In animal studies, CTLA-4 blockade is associated with higher incidence of pregnancy loss. IgG2 is known to cross the placental barrier. Tremelimumab is not recommended during pregnancy and in women of childbearing potential not using effective contraception during treatment and for at least 3 months after the last dose.
In reproduction studies, intravenous administration of tremelimumab at doses up to 30 mg/kg/week to pregnant cynomolgus monkeys during the period of organogenesis was not associated with maternal toxicity or effects, pregnancy losses, fetal weights, or external, visceral, skeletal abnormalities or weights of selected fetal organs at clinically relevant exposures.
There is no information regarding the presence of tremelimumab in human milk, the absorption and effects on the breastfed infant, or the effects on milk production. IgG2 is excreted in human milk. Because of the potential for adverse reactions from tremelimumab in breastfed infants, lactating women are advised not to breastfeed during treatment and for at least 3 months after the last dose.4.7 Effects on Ability to Drive and Use Machines
Based on its pharmacodynamic properties, tremelimumab is unlikely to affect the ability to drive and use machines. However, if patients experience adverse reactions affecting their ability to concentrate and react, they should be advised to use caution when driving or operating machinery.
4.8 Adverse Effects (Undesirable Effects)
Clinical trials experience.
The safety of STRIDE is based on data in 462 patients from the HIMALAYA study and Study 22 (uHCC, HCC pool).
HIMALAYA study.
The safety of Imjudo administered in combination with durvalumab was evaluated in a total of 388 patients with uHCC in HIMALAYA, a randomised, open-label, multicentre study (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Patients received Imjudo 300 mg administered as a single intravenous infusion in combination with durvalumab 1,500 mg on the same day, followed by durvalumab every 4 weeks or sorafenib 400 mg given orally twice daily.
Serious adverse events occurred in 41% of patients who received Imjudo in combination with durvalumab. Serious adverse events in > 1% of patients included haemorrhage (6%), diarrhoea (4%), sepsis (2.1%), pneumonia (2.1%), rash (1.5%), vomiting (1.3%), acute kidney injury (1.3%), and anaemia (1.3%). Fatal adverse events occurred in 8% of patients who received Imjudo in combination with durvalumab, including death (1%), haemorrhage intracranial (0.5%), cardiac arrest (0.5%), pneumonitis (0.5%), hepatic failure (0.5%), and immune-mediated hepatitis (0.5%). The most common adverse events (occurring in ≥ 20% of patients) were rash, diarrhoea, fatigue, pruritus, musculoskeletal pain, and abdominal pain.
Permanent discontinuation of the treatment regimen due to an adverse event occurred in 14% of patients; the most common adverse events leading to treatment discontinuation (≥ 1%) were haemorrhage (1.8%), diarrhoea (1.5%), AST increased (1%), and hepatitis (1%).
Dosage interruptions or delay of the treatment regimen due to an adverse event occurred in 35% of patients. Adverse events which required dosage interruption or delay in ≥ 1% of patients included ALT increased (3.6%), diarrhoea (3.6%), rash (3.6%), amylase increased (3.4%), AST increased (3.1%), lipase increased (2.8%), pneumonia (1.5%), hepatitis (1.5%), pyrexia (1.5%), anaemia (1.3%), thrombocytopenia (1%), hyperthyroidism (1%), pneumonitis (1%), and blood creatinine increased (1%).
Table 3 summarises the adverse events that occurred in patients treated with STRIDE in the HIMALAYA study.
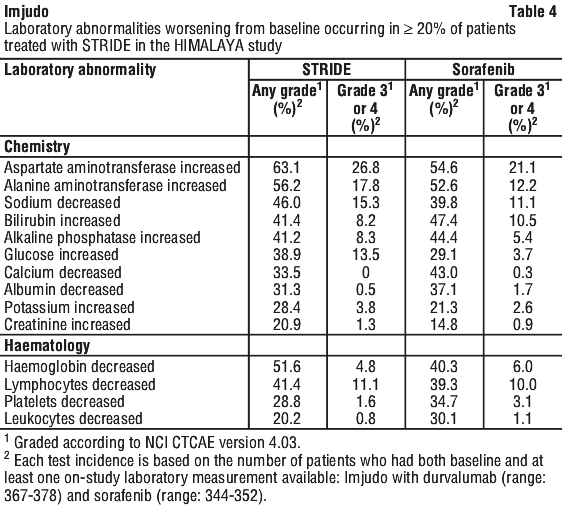
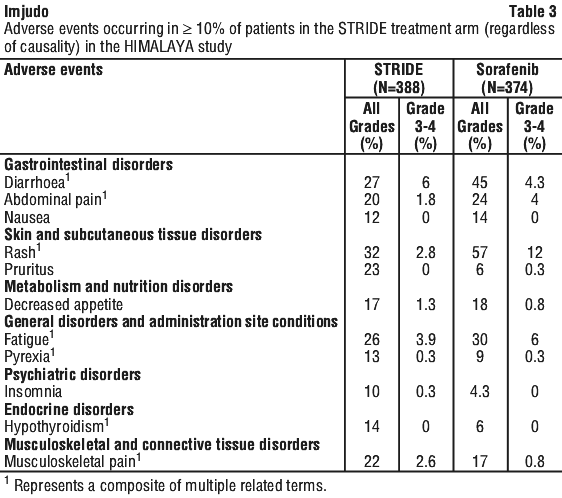 Table 4 summarises the laboratory abnormalities that occurred in patients treated with STRIDE in the HIMALAYA study.
Table 4 summarises the laboratory abnormalities that occurred in patients treated with STRIDE in the HIMALAYA study.
 The safety of STRIDE is based on data in 462 patients from the HCC pool (uHCC) and was consistent with known Imjudo + durvalumab safety profile.
The safety of STRIDE is based on data in 462 patients from the HCC pool (uHCC) and was consistent with known Imjudo + durvalumab safety profile.
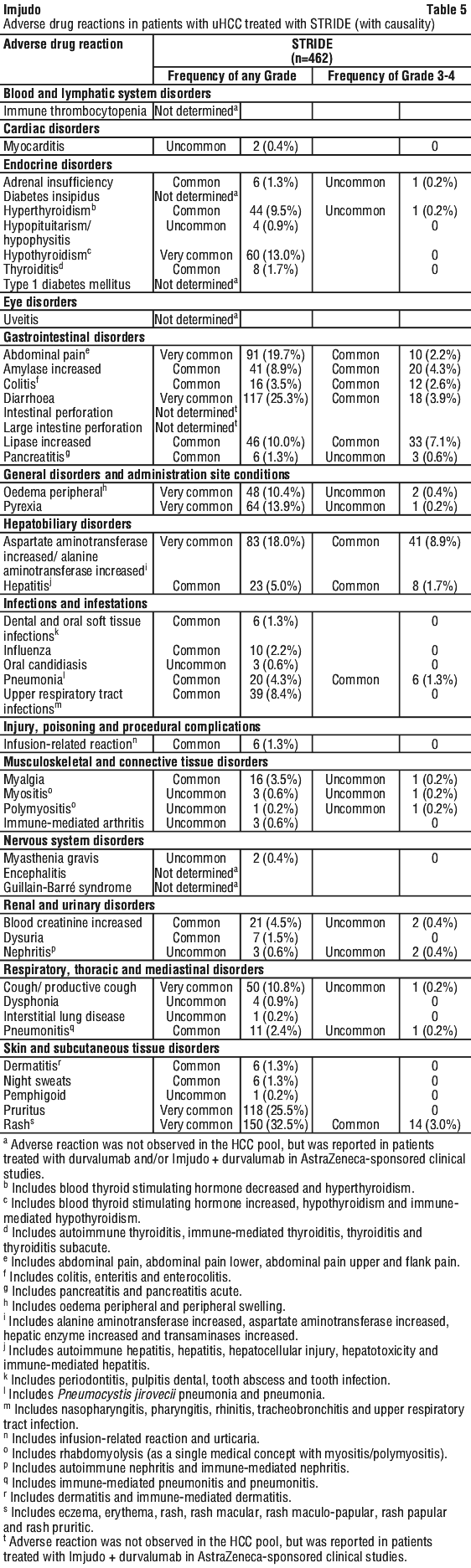
Tabulated list of adverse reactions.
Table 5 lists the incidence of ADRs in patients treated with STRIDE in the HCC pool. Adverse drug reactions are listed according to system organ class in MedDRA. Within each system organ class, the adverse drug reactions are presented in decreasing frequency. Within each frequency grouping, ADRs are presented in order of decreasing seriousness. In addition, the corresponding frequency category for each ADR is based on the CIOMS III convention and is defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1000); very rare (< 1/10,000); not determined (cannot be estimated from available data).
Description of selected adverse reactions.
The data below reflects information for significant adverse reactions for STRIDE in the HCC pool (n=462).
The management guidelines for these adverse reactions are described in Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use.
Immune-mediated pneumonitis. In patients receiving STRIDE, immune-mediated pneumonitis occurred in 6 (1.3%) patients, including Grade 3 in 1 (0.2%) patient and Grade 5 (fatal) in 1 (0.2%) patient. The median time to onset was 29 days (range: 5-774 days). All patients received systemic corticosteroids, and 5 of the 6 patients received high-dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). One patient also received other immunosuppressants. Treatment was discontinued in 2 patients. Resolution occurred in 3 patients.
Immune-mediated hepatitis. In patients receiving STRIDE, immune mediated hepatitis occurred in 34 (7.4%) patients, including Grade 3 in 20 (4.3%) patients, Grade 4 in 1 (0.2%) patient and Grade 5 (fatal) in 3 (0.6%) patients. The median time to onset was 29 days (range: 13-313 days). All patients received systemic corticosteroids, and 32 of the 34 patients received high dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). Nine patients also received other immunosuppressants. Treatment was discontinued in 10 patients. Resolution occurred in 13 patients.
Immune-mediated colitis. In patients receiving STRIDE, immune mediated colitis or diarrhoea occurred in 31 (6.7%) patients, including Grade 3 in 17 (3.7%) patients. The median time to onset was 23 days (range: 2-479 days). All patients received systemic corticosteroids, and 28 of the 31 patients received high dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). Four patients also received other immunosuppressants. Treatment was discontinued in 5 patients. Resolution occurred in 29 patients.
Intestinal perforation was not observed in patients receiving STRIDE.
Immune-mediated endocrinopathies. Immune-mediated hypothyroidism.
In patients receiving STRIDE, immune-mediated hypothyroidism occurred in 46 (10.0%) patients. The median time to onset was 85 days (range: 26-763 days). One patient received high-dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). All patients required other therapy (thiamazole, carbimazole, propylthiouracil, perchlorate, calcium channel blocker, or beta-blocker). Resolution occurred in 6 patients. Immune-mediated hypothyroidism was preceded by immune-mediated hyperthyroidism in 4 patients.
Immune-mediated hyperthyroidism.
In patients receiving STRIDE, immune-mediated hyperthyroidism occurred in 21 (4.5%) patients, including Grade 3 in 1 (0.2%) patient. The median time to onset was 30 days (range: 13-60 days). Four patients received systemic corticosteroids, and all of the four patients received high-dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). Twenty patients required other therapy (thiamazole, carbimazole, propylthiouracil, perchlorate, calcium channel blocker, or beta-blocker). One patient discontinued treatment due to hyperthyroidism. Resolution occurred in 17 patients.
Immune-mediated thyroiditis.
In patients receiving STRIDE, immune-mediated thyroiditis occurred in 6 (1.3%) patients. The median time to onset was 56 days (range: 7-84 days). Two patients received systemic corticosteroids, and 1 of the 2 patients received high-dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). All patients required other therapy including hormone replacement therapy, thiamazole, carbimazole, propylthiouracil, perchlorate, calcium channel blocker, or beta-blocker. Resolution occurred in 2 patients.
Immune-mediated adrenal insufficiency.
In patients receiving STRIDE, immune-mediated adrenal insufficiency occurred in 6 (1.3%) patients, including Grade 3 in 1 (0.2%) patient. The median time to onset was 64 days (range: 43-504 days). All patients received systemic corticosteroids, and 1 of the 6 patients received high-dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). Resolution occurred in 2 patients.
Immune-mediated type 1 diabetes mellitus.
In patients receiving STRIDE, immune-mediated type 1 diabetes mellitus was not observed.
Immune-mediated hypophysitis/hypopituitarism.
In patients receiving STRIDE, immune-mediated hypophysitis/hypopituitarism occurred in 5 (1.1%) patients. The median time to onset for the events was 149 days (range: 27-242 days). Four patients received systemic corticosteroids, and 1 of the 4 patients received high-dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). Three patients also required endocrine therapy. Resolution occurred in 2 patients.
Immune-mediated nephritis. In patients receiving STRIDE, immune-mediated nephritis occurred in 4 (0.9%) patients, including Grade 3 in 2 (0.4%) patients. The median time to onset was 53 days (range: 26-242 days). All patients received systemic corticosteroids, and 3 of the 4 received high-dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). Treatment was discontinued in 2 patients. Resolution occurred in 3 patients.
Immune-mediated rash. In patients receiving STRIDE, immune-mediated rash or dermatitis (including pemphigoid) occurred in 26 (5.6%) patients, including Grade 3 in 9 (1.9%) patients and Grade 4 in 1 (0.2%) patients. The median time to onset was 25 days (range: 2-933 days). All patients received systemic corticosteroids and 14 of the 26 patients received high-dose corticosteroid treatment (at least 40 mg prednisone or equivalent per day). One patient received other immunosuppressants. Treatment was discontinued in 3 patients. Resolution occurred in 19 patients.
Infusion-related reactions. In patients receiving STRIDE, infusion-related reactions occurred in 7 (1.5%) patients.
Post-marketing experience.
Adverse drug reactions in patients treated with STRIDE. Gastrointestinal disorders.
Coeliac disease.
Musculoskeletal and connective tissue disorders.
Sjögren's syndrome, tenosynovitis, polymyalgia rheumatica.
Immune checkpoint inhibitor class effects. There have been cases of the following adverse reactions reported during treatment with other immune checkpoint inhibitors which might also occur during treatment with STRIDE: haemophagocytic lymphohistiocytosis (HLH), pancreatic exocrine insufficiency and aplastic anaemia.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no specific treatment in the event of tremelimumab overdose, and symptoms of overdose are not established. In the event of an overdose, physicians should follow general supportive measures and should treat symptomatically.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
CTLA-4 is primarily expressed on the surface of T lymphocytes. Interaction of CTLA-4 with its ligands, CD80 and CD86, limits effector T-cell activation, through a number of potential mechanisms, but primarily by limiting co-stimulatory signalling through CD28.
Tremelimumab is a selective, fully human IgG2 antibody that blocks CTLA-4 interaction with CD80 and CD86, thus enhancing T-cell activation and proliferation, resulting in increased T-cell diversity and enhanced antitumour immune activity. In syngeneic mouse tumour models, blocking CTLA-4 activity resulted in decreased tumour growth and increased proliferation of T cells in tumours.
The combination of durvalumab, a PD-L1 inhibitor, and tremelimumab functions to enhance anti-tumour T-cell activation and functions at multiple stages of the immune response, maximizing anti-tumour immunity.
The effect of STRIDE on the quantities of proliferative cytotoxic CD8+ T cells was evaluated in Study 22 in patients with uHCC using a CD8+Ki67+ assay. At Day 15 a marked increase of proliferating CD8+ T cell populations was observed in the STRIDE arm compared to the durvalumab monotherapy arm. Patients receiving STRIDE also experienced a higher Objective Response Rate (ORR) compared to other treatment arms and responders across all arms exhibited higher median proliferative cytotoxic CD8+ T cell when compared to non-responding patients.
Clinical trials.
HCC - HIMALAYA study.
The efficacy of STRIDE was evaluated in the HIMALAYA study, a randomised, open-label, multicentre study in patients with confirmed uHCC who did not receive prior systemic treatment for HCC. The study included patients with BCLC Stage C or B (not eligible for locoregional therapy) and Child-Pugh Score Class A.
The study excluded patients with co-infection of viral hepatitis B and hepatitis C; active or prior documented GI bleeding within 12 months; ascites requiring non-pharmacologic intervention within 6 months; hepatic encephalopathy within 12 months before the start of treatment; active or prior documented autoimmune or inflammatory disorders.
Patients with oesophageal varices were included except those with active or prior documented GI bleeding within 12 months prior to study entry.
Randomisation was stratified by macrovascular invasion (MVI) (yes vs. no), aetiology of liver disease (confirmed hepatitis B virus vs. confirmed hepatitis C virus vs. others) and ECOG performance status (0 vs. 1).
The HIMALAYA study randomized 1171 patients 1:1:1 to receive:
D: durvalumab 1500 mg every 4 weeks;
STRIDE: Imjudo 300 mg as a single priming dose + durvalumab 1500 mg; followed by durvalumab 1500 mg every 4 weeks;
S: Sorafenib 400 mg twice daily.
Treatment continued as long as clinical benefit was observed or until unacceptable toxicity. Patients in all arms could continue to receive treatment after evidence of disease progression if, in the Investigator's opinion, they were benefiting from study drug and met all inclusion and exclusion criteria for treatment beyond progression. In addition, patients in the STRIDE arm who continued treatment beyond progression were allowed to be rechallenged once with an additional single dose of Imjudo 300 mg after cycle five of durvalumab. Of the 182 patients enrolled to the STRIDE arm who received durvalumab beyond progression, the median OS was 19.5 months (95% CI: 15.4, 23.4). Of the 30 patients who were enrolled to the STRIDE arm who were rechallenged with Imjudo, the median OS was 30.4 months (95% CI: 23.4, NR).
Tumour assessments were conducted every 8 weeks for the first 12 months and then every 12 weeks thereafter. Survival assessments were conducted every month for the first 3 months following treatment discontinuation and then every 2 months.
The primary endpoint was OS. Key secondary endpoints were PFS, Investigator assessed ORR and DoR according to RECIST v1.1. Patient-Reported Outcomes (PROs) were also assessed.
The demographics and baseline disease characteristics were generally representative for patients with uHCC. The baseline demographics of the overall study population were as follows: male (83.7%), age < 65 years (50.4%), white (44.6%), Asian (50.7%), black or African American (1.7%), other (2.3%), ECOG PS 0 (62.6%); Child-Pugh Class score A (99.5%), macrovascular invasion (25.2%), extrahepatic spread (53.4%), viral aetiology; hepatitis B (30.6%), hepatitis C (27.2%), uninfected (42.2%).
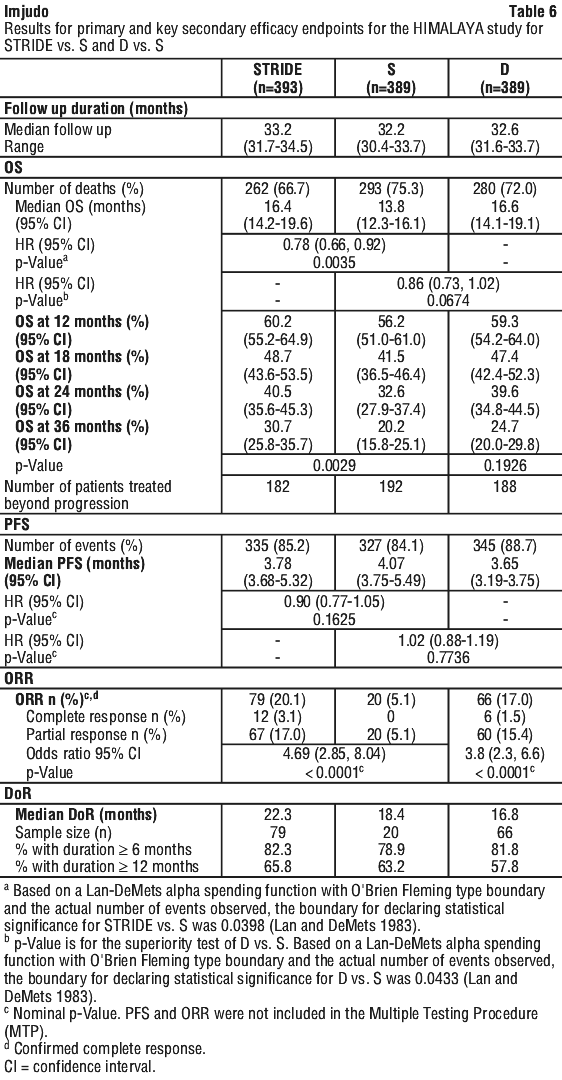
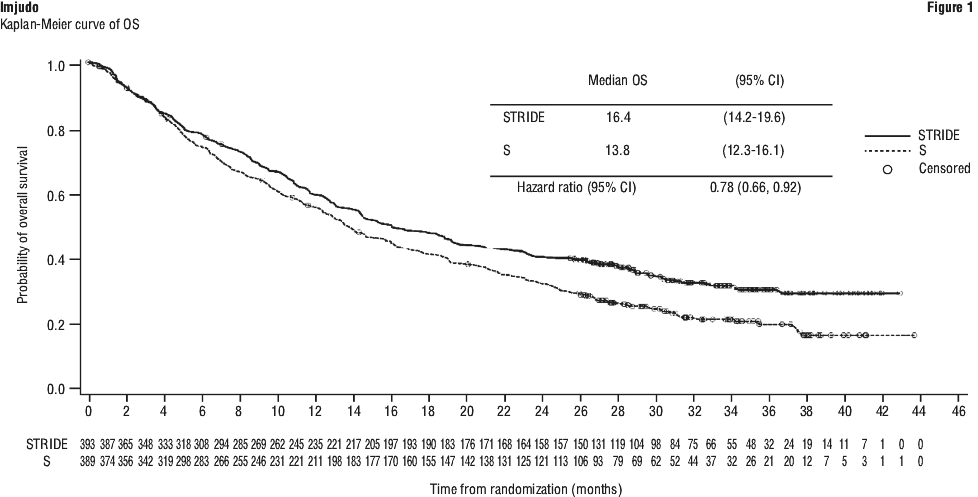
The study demonstrated a statistically significant and clinically meaningful improvement in OS with STRIDE vs. S [HR = 0.78 [95% CI 0.66, 0.92]; p = 0.0035].
See Table 6 and Figure 1.
 Patient reported outcomes. Patient-reported symptoms, function and health-related quality of life (HRQoL) were collected using the EORTC QLQ-C30 and its hepatocellular carcinoma module (EORTC QLQ-HCC18).
Patient reported outcomes. Patient-reported symptoms, function and health-related quality of life (HRQoL) were collected using the EORTC QLQ-C30 and its hepatocellular carcinoma module (EORTC QLQ-HCC18).
At baseline, patient-reported symptoms, functioning or HRQoL scores were comparable between the study arms.
Delay in time to deterioration of symptoms, functioning, and global health status/QoL.
STRIDE vs. S demonstrated a clinically meaningful improvement by delaying time to deterioration in a broad range of patient-reported symptoms, function, and global health status/QoL compared to S. Longer time to deterioration (median in months) was observed in the STRIDE arm compared to S for the following symptoms: Global Health Status (7.5 vs. 5.7 months, HR 0.76, p = 0.0306); physical functioning (12.9 vs. 7.4 months, HR 0.68; p = 0.0020), fatigue (7.4 vs. 5.4 months, HR 0.71; p = 0.0026), nausea (25.0 vs. 11.0 months, HR 0.65; p = 0.0033), appetite loss (12.6 vs. 6.9 months, HR 0.59; p < 0.0001), abdominal pain (16.8 vs. 8.9 months, HR 0.61; p = 0.0008) and abdominal swelling (20.9 vs. 11.1 months, HR 0.74; p = 0.0431.
Change from baseline in patient-reported symptoms (mixed model for repeated measures).
STRIDE improved patient-reported HRQoL functioning and diarrhoea by demonstrating a nominal difference and clinically meaningful mean change from baseline vs. S from randomisation until 8 months (estimated mean difference at 8 months: -18.5 95% CI: -23.24, -13.84 and p-Value: < 0.0001).
Patient-reported outcome results should be interpreted in the context of the open-label study design.
HCC - Study 22.
The safety and efficacy of STRIDE was evaluated in Study 22, an open-label, uncontrolled, multi-part Phase I/II, study involving 433 immunotherapy-naïve patients with uHCC. Of the 75 patients who received proposed combination treatment regimen T300+D, more than a quarter received T300+D as first line of systemic therapy (73.3% had received prior systemic therapy with sorafenib/ other VEGFR TKI). The higher percentage of patients alive and in survival follow-up (including those still receiving study treatment) in the T300+D arm (30.7%) compared to the other treatment arms (17.4% to 19.2%) at DCO was more likely indicative of the data in the T300+D arm being less mature than that in the other 3 arms as enrolment in the T300+D safety run-in arm (Part 2B) began approximately 8 months after the start of the other 3 treatment arms (in Part 2A).
The study included patients with BCLC Stage C or B (not eligible for locoregional therapy), ECOG performance status of 0 or 1 and Child-Pugh Score Class A.
The study excluded patients with co-infection of viral hepatitis B and hepatitis C; active or prior documented GI bleeding within 12 months; ascites requiring non-pharmacologic intervention within 6 months; hepatic encephalopathy within 12 months before the start of treatment; active or prior documented autoimmune or inflammatory disorders.
Treatment continued as long as clinical benefit was observed or until unacceptable toxicity. Patients who completed the assigned dosing cycles and were benefiting from study drug in the Investigator's opinion and subsequently had evidence of disease progression during the durvalumab monotherapy phase could be rechallenged with Imjudo 300 mg.
Tumour assessments were conducted every 8 weeks.
The primary objective was safety and tolerability. Key secondary endpoints included OS, ORR and DoR. ORR and DoR were based on Investigator assessments and BICR according to RECIST 1.1.
The baseline demographics of the study population (STRIDE) were as follows: male (86.7%); age <65 years (45.3%), white (36.0%); Asian (58.7%); black or African American (5.3%); other (0%), ECOG PS 0 (61.3%), Child-Pugh Class/Score A/5 (68.0%), Child-Pugh Class/Score A/6 (30.7%), macrovascular invasion (21.3%); extrahepatic spread (70.7%), viral aetiology; hepatitis B (36.0%), hepatitis C (28.0%), uninfected (36.0%); prior systemic therapy (73.3%).
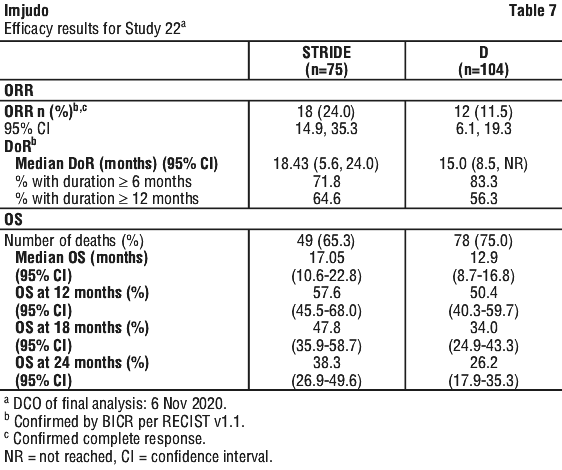
Efficacy results are shown in Table 7.
5.2 Pharmacokinetic Properties
The pharmacokinetics of tremelimumab as a single agent was studied in patients with doses ranging from 75 mg to 750 mg or 10 mg/kg administered once every 4 or 12 weeks, and in combination with durvalumab at a single priming dose of 300 mg.
PK exposure increased dose proportionally at doses ≥ 75 mg every 4 weeks. Steady state was achieved at approximately 12 weeks.
The pharmacokinetics of tremelimumab was similar when assessed as a single agent and when in combination with durvalumab.
Distribution.
From a final two-compartmental tremelimumab population PK model, the geometric mean (% coefficient of variation [CV%]) for central (V1) and peripheral (V2) volume of distribution was 3.45 (27.3%) and 2.47 (43.3%) L, respectively.
Excretion.
Tremelimumab clearance decreases over time, with a mean maximal reduction (CV%) from baseline values of approximately 22.7% (26.1%) resulting in a geometric mean (CV%) steady state clearance (CLss) of 0.202 L/day (19.2%); the decrease in CLss is not considered clinically relevant. The geometric mean (CV%) terminal half-life was approximately 20.4 (34.7%) days.
Special populations.
Age (18-87 years), body weight (34-149 kg), gender, positive anti-drug antibody (ADA) status, albumin levels, LDH levels, creatinine levels, tumour type, race, mild renal impairment (creatinine clearance (CRCL) 60 to 89 mL/min), moderate renal impairment (creatinine clearance (CRCL) 30 to 59 mL/min), mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin > 1.0 to 1.5 x ULN and any AST), moderate hepatic impairment (bilirubin > 1.5 to 3 x ULN and any AST) or ECOG/WHO status had no clinically significant effect on the PK of tremelimumab.
The effect of severe renal impairment (CRCL 15 to 29 mL/min) or severe hepatic impairment (bilirubin > 3.0 x ULN and any AST) on the PK of tremelimumab is unknown.
Immunogenicity.
As with all therapeutic proteins, there is a potential for immunogenicity. Immunogenicity of tremelimumab is based on pooled data in 2075 patients who were treated with Imjudo 75 mg or 1 mg/kg and evaluable for the presence of anti-drug antibodies (ADAs). Two-hundred fifty-two patients (12.1%) tested positive for treatment-emergent ADAs. Neutralizing antibodies against tremelimumab were detected in 10.0% (208/2075) patients. The presence of ADAs did not impact tremelimumab pharmacokinetics, and there was no apparent effect on efficacy and safety.
In the HIMALAYA study, of the 182 patients who were treated with STRIDE and evaluable for the presence of ADAs against tremelimumab, 20 (11.0%) patients tested positive for treatment-emergent ADAs. Neutralizing antibodies against tremelimumab were detected in 4.4% (8/182) patients. The presence of ADAs did not have an apparent effect on pharmacokinetics or safety.
Immunogenicity assay results are highly dependent on several factors, including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease.
For these reasons, comparison of incidence of antibodies to tremelimumab with the incidence of antibodies to other products may be misleading.
5.3 Preclinical Safety Data
Genotoxicity.
The genotoxic potential of tremelimumab has not been evaluated.
Carcinogenicity.
The carcinogenic potential of tremelimumab has not been evaluated.6 Pharmaceutical Particulars
6.1 List of Excipients
Histidine, histidine hydrochloride monohydrate, trehalose dihydrate, disodium edetate, polysorbate 80 and water for injections.
6.2 Incompatibilities
No incompatibilities between Imjudo and 9 g/L (0.9%) sodium chloride or 50 g/L (5%) dextrose in polyvinylchloride or polyolefin IV bags have been observed.
This drug product must not be mixed with other drug products except those mentioned in Section 4.2 Dose and Method of Administration.
Do not co-administer other drugs through the same intravenous line.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store unopened vials under refrigeration at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not freeze. Do not shake.
6.5 Nature and Contents of Container
1.25 mL (a total of 25 mg tremelimumab) concentrate in a Type I glass vial with an elastomeric stopper and a violet flip-off aluminum seal. Pack size of 1 single-dose vial.
15 mL (a total of 300 mg tremelimumab) concentrate in a Type I glass vial with an elastomeric stopper and a dark blue flip-off aluminum seal. Pack size of 1 single-dose vial.
Not all pack sizes and strengths may be available in Australia.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Imjudo is a human anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4)- immunoglobulin G2 (IgG2a) monoclonal antibody.
CAS number.
745013-59-6.7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Summary Table of Changes
