What is in this leaflet
This leaflet answers some common questions about Inspra.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Inspra against the benefits it is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Inspra is used for
What Inspra does
- This medicine is used to treat heart failure in patients who have experienced a heart attack.
A heart attack occurs when one of the major blood vessels supplying blood to your heart becomes blocked. This means that your heart cannot receive the oxygen it needs and becomes damaged. This may lead to further problems, such as heart failure, irregular heart rhythms and blood clots.
Heart failure means that the heart muscle is weak and cannot pump blood strongly enough to supply all the blood needed throughout the body. Heart failure is not the same as heart attack, and may start off with mild or no symptoms, but as the condition progresses, patients may feel short of breath or may get tired easily after light physical activity such as walking. Some patients may wake up short of breath at night, or have to prop their heads up during sleep to avoid this problem. Fluid may collect in different parts of the body, often first noticed as swollen ankles and feet. - This medicine is also used to reduce the risk of death or need for hospitalisations due to heart failure in patients with chronic heart failure.
How Inspra works
Your body makes a substance called aldosterone. It is important for regulating blood pressure and is one of the factors involved in heart function. Sometimes aldosterone can cause changes in our body that lead to heart failure. Inspra works by blocking the action of aldosterone, and slowing the progression of heart failure by reducing heart damage.
Inspra belongs to a group of medicines called 'selective aldosterone blockers' that stop the action of aldosterone.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
There is no evidence that this medicine is addictive.
This medicine is available only with a doctor's prescription.
Use in children
The safety and effectiveness of Inspra in children have not been established.
Before you take Inspra
When you must not take it
Do not take Inspra if you have an allergy to:
- any medicine containing eplerenone
- any medicine containing any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- skin rash
- itchiness
- shortness of breath
- swelling of the face, lips or tongue
- muscle pain or tenderness
- joint pain.
Do not take Inspra if you have or have had any of the following medical conditions:
- very high levels of potassium in your blood
- severely reduced kidney function. Your doctor will determine your kidney function.
- Severe liver problems.
Do not take Inspra if you are currently taking any of the following medicines:
- potassium-sparing diuretics (e.g. spironolactone, amiloride), used to help the kidneys get rid of salt and water by increasing the amount of urine produced
- ketoconazole and itraconazole used to treat fungal infections
- clarithromycin, used to treat bacterial infections
- saquinavir, ritonavir, for the treatment of HIV infections.
Taking Inspra together with the above medicines can lead to serious side effects.
Do not take this medicine if you are pregnant. It may affect your developing baby if you take it during pregnancy.
Do not breast-feed if you are taking this medicine. The active ingredient in Inspra may pass into breast milk and there is a possibility that your baby may be affected.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- high levels of potassium in your blood
- diabetes
- long term kidney disease
- liver problems.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell them before you start taking Inspra.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including:
- all prescription medicines
- all medicines you buy over the counter from a pharmacy or supermarket
- all complementary and alternative therapies
- anything you buy from a health food shop.
Some medicines may be affected by Inspra or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines. Your doctor will advise you.
Tell your doctor or pharmacist if you are taking any of the following:
- medicines known as angiotensin converting enzyme (ACE) inhibitor and/or angiotensin receptor blocker (ARB), such as quinapril, losartan. These are used to treat high blood pressure and some other heart conditions and may increase the risk of high potassium levels in your blood.
- non-steroidal anti-inflammatory drugs (NSAIDs), medicines used to relieve pain, swelling and other symptoms of inflammation including aspirin and ibuprofen.
- lithium, a medicine used to treat mood swings
- neuroleptics, used to treat certain mental illnesses
- tricyclic antidepressants, used to treat certain mental illnesses
- St John's Wort, used in the management of depression
- carbamazepine, used to control seizures, facial pain or certain types of mood disorders
- phenytoin and phenobarbitone, medicines used to control seizures
- potassium-sparing diuretics, such as spironolactone, amiloride
- potassium supplements, or salt substitutes which contain potassium
- medicines used to treat fungal infections such as ketoconazole, itraconazole
- certain antibiotics used to treat bacterial infections, such as erythromycin, trimethoprim, rifampicin
- saquinavir, ritonavir, for the treatment of HIV infections
- immunosupressive agents such as cyclosporin, tacrolimus
- baclofen, a muscle relaxant
- prazosin, used to treat high blood pressure and other medical conditions
- alfuzosin, for the treatment of benign prostatic hyperplasia
- amifostine, used in combination with cancer treatments
- any other medicines used to treat high blood pressure or heart failure.
If you are not sure if you are taking any of these medicines mentioned in this leaflet, check with your doctor or pharmacist.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
Tell your doctor if you are taking salt tablets. Taking Inspra together with salt tablets can lead to serious side effects.
How to take Inspra
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how many tablets you need to take each day. This may depend upon your age, your kidney condition, the potassium level in your blood, and whether or not you are taking any other medicines.
The usual starting dose of Inspra is 25 mg taken once a day. After about 4 weeks, your doctor may increase the dose to 50 mg once a day.
Your doctor will do blood tests to help determine the correct dose of Inspra for you.
How to take it
Swallow the tablets whole with a full glass of water.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take the tablets.
Your tablets may be taken with or after a meal, or on an empty stomach.
How long to take it
Continue taking your medicine for as long as your doctor tells you. This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it
If it is less than 12 hours before your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Australian Poisons Information Centre (telephone 13 11 26) or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Inspra. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you take too much Inspra, you may feel light-headed.
While you are taking Inspra
Things you must do
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may occasionally do a blood test to check your potassium levels and see how your kidneys are working. Your dose of Inspra may be adjusted by your doctor, depending on the potassium levels in your blood.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Inspra.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor that you are taking this medicine.
Make sure you drink enough water during exercise and hot weather when you are taking this medicine, especially if you sweat a lot. If you do not drink enough water while taking Inspra, you may feel faint, light-headed or sick. This is because your blood pressure is dropping suddenly. If you continue to feel unwell, tell your doctor.
If you have excess vomiting or diarrhoea while taking Inspra, tell your doctor. You may lose too much water and salt and your blood pressure may drop too much.
If you feel light-headed or dizzy after taking your first dose of Inspra, or when your dose is increased, tell your doctor immediately.
Things you must not do
Do not take Inspra to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine, or lower the dosage, without checking with your doctor. If you stop taking it suddenly, your condition may worsen or you may have unwanted side effects.
Things to be careful of
Be careful driving or operating machinery until you know how Inspra affects you. This medicine may cause dizziness and feeling faint in some people. If you have this symptom, do not drive, operate machinery or do anything else that could be dangerous.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Inspra or your condition changes. Tell your doctor even if you think the problem is not connected with the medicine or is not listed in this leaflet.
This medicine helps most people with heart failure, but it may have unwanted side effects in some people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
It is often difficult to tell whether side effects are the result of taking Inspra, or the effects of your heart failure or side effects of other medicines you may be taking. For this reason it is important to report any change in your condition. Your doctor may want to change your dose or advise you to stop taking Inspra.
If you are over 65 years of age you may have an increased chance of having some side effects, as you may be more sensitive to the effects of the medication.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if...
The following list includes the more common or noticeable side effects of your medicine.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- feeling light-headed, dizzy or faint
- stomach or bowel problems
- feeling sick (nausea) or vomiting
- diarrhoea
- constipation
- flatulence or wind - cough
- sore throat
- headache
- rash, itchy skin
- high temperature, signs of an infection
- back pain.
Tell your doctor as soon as possible if...
The following list includes side effects that may require medical attention. Serious side effects are rare.
Tell your doctor as soon as possible if you notice any of the following:
- heart flutters, increased heart rate
- unusual tiredness, weakness
- muscle spasms and pain
- abdominal pain
- enlargement of the breasts in men
- reduced sense of touch
- increased sweating
- feeling weak and generally unwell
- problems with sleeping.
Go to hospital if...
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital, if you notice any of the following:
- shortness of breath, swelling of the feet or legs due to fluid build up
- chest pain which may spread to the neck and shoulders
- swelling of the face, lips, mouth, tongue or throat which may cause difficulty in swallowing or breathing.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some patients.
Some of these side effects (for example, changes in potassium levels, thyroid function, or cholesterol level) can only be found when your doctor does tests from time to time to check your progress.
After taking Inspra
Storage
Keep your Inspra tablets in the pack until it is time to take them. If you take the tablets out of the pack they will not keep well.
Keep your tablets in a cool dry place where the temperature stays below 25 °C.
Do not store Inspra or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Inspra is a yellow, arc diamond, film-coated tablet.
Packs contain 30 tablets.
25 mg tablet
Marked with 'NSR' over '25' on one side and 'Pfizer' on the other.
50 mg tablet
Marked with 'NSR' over '50' on one side and 'Pfizer' on the other.
Ingredients
The active ingredient in Inspra is eplerenone.
- Inspra 25 tablet contains 25 mg eplerenone
- Inspra 50 tablet contains 50 mg eplerenone.
Inspra tablets also contain:
- lactose
- microcrystalline cellulose
- croscarmellose sodium
- hydromellose
- sodium lauryl sulfate
- talc-purified
- magnesium stearate
- titanium dioxide
- macrogol 400
- polysorbate 80
- iron oxide yellow (CI77492)
- iron oxide red (CI77491).
This medicine does not contain sucrose, gluten, or tartrazine.
Supplier
Inspra is supplied in Australia by:
Viatris Pty Ltd
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
Australian registration numbers
25 mg AUST R 100162
50 mg AUST R 100163
This leaflet was prepared in August 2021.
INSPRA® is a Viatris company trade mark
ujcinspt10821
Published by MIMS December 2021
 Inspra should be suspended when serum potassium is ≥ 6.0 mmol/L. It can be restarted at a dose of 25 mg every other day when serum potassium levels have fallen below 5.0 mmol/L. Serum potassium monitoring should continue once eplerenone has been restarted again.
Inspra should be suspended when serum potassium is ≥ 6.0 mmol/L. It can be restarted at a dose of 25 mg every other day when serum potassium levels have fallen below 5.0 mmol/L. Serum potassium monitoring should continue once eplerenone has been restarted again. Doses above 25 mg daily have not been studied in patients with eGFR < 50 mL/min/1.73 m2.
Doses above 25 mg daily have not been studied in patients with eGFR < 50 mL/min/1.73 m2. See Tables 4 and 5.
See Tables 4 and 5.
 A total of 3,353 patients have been treated with Inspra in clinical studies of hypertension. The overall rates of adverse events in placebo controlled studies were similar between Inspra (49%) and placebo (48%). Adverse events with suspected relationship to treatment and in excess of placebo from the monotherapy arms of five placebo controlled studies for patients who received Inspra 25 to 400 mg are listed below by absolute frequency. Frequencies are defined as common (> 1% to ≤ 10%) or uncommon (> 0.1% to ≤ 1%).
A total of 3,353 patients have been treated with Inspra in clinical studies of hypertension. The overall rates of adverse events in placebo controlled studies were similar between Inspra (49%) and placebo (48%). Adverse events with suspected relationship to treatment and in excess of placebo from the monotherapy arms of five placebo controlled studies for patients who received Inspra 25 to 400 mg are listed below by absolute frequency. Frequencies are defined as common (> 1% to ≤ 10%) or uncommon (> 0.1% to ≤ 1%). Table 7 shows the rates of hyperkalaemia in EPHESUS as assessed by baseline renal function (creatinine clearance).
Table 7 shows the rates of hyperkalaemia in EPHESUS as assessed by baseline renal function (creatinine clearance). Table 8 shows the rates of hyperkalaemia in EPHESUS as assessed by two baseline characteristics: presence/ absence of proteinuria from baseline urinalysis and presence/ absence of diabetes (see Section 4.4 Special Warnings and Precautions for Use, Hyperkalaemia).
Table 8 shows the rates of hyperkalaemia in EPHESUS as assessed by two baseline characteristics: presence/ absence of proteinuria from baseline urinalysis and presence/ absence of diabetes (see Section 4.4 Special Warnings and Precautions for Use, Hyperkalaemia).

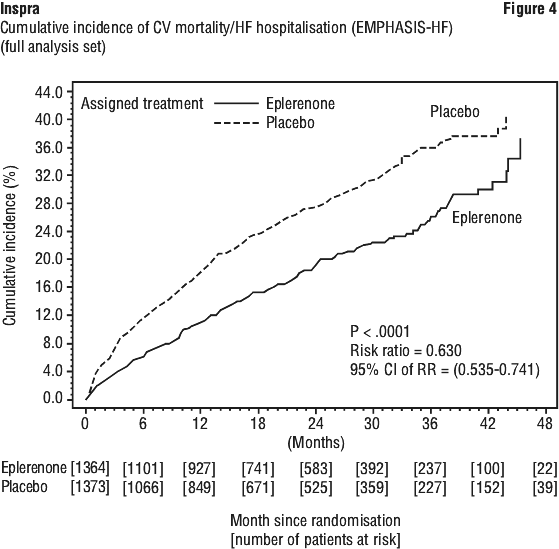
 The reduction in mortality observed in patients treated with Inspra compared to those who received placebo is mainly the result of a reduction in the rate of sudden death after myocardial infarction. In the first 12 months of treatment the rate of all cause mortality was 11.68% among patients treated with Inspra compared to 13.63% for patients treated with placebo. Among patients who remained alive after 12 months of therapy, the all-cause mortality rates at month 27 in the eplerenone and placebo groups were 7.97% and 9.58%, respectively.
The reduction in mortality observed in patients treated with Inspra compared to those who received placebo is mainly the result of a reduction in the rate of sudden death after myocardial infarction. In the first 12 months of treatment the rate of all cause mortality was 11.68% among patients treated with Inspra compared to 13.63% for patients treated with placebo. Among patients who remained alive after 12 months of therapy, the all-cause mortality rates at month 27 in the eplerenone and placebo groups were 7.97% and 9.58%, respectively. Analyses conducted for a variety of CV biomarkers did not confirm a mechanism of action by which mortality was reduced.
Analyses conducted for a variety of CV biomarkers did not confirm a mechanism of action by which mortality was reduced.
 In dose ranging studies of chronic heart failure (NYHA classification II-IV), the addition of eplerenone to standard therapy resulted in expected dose dependent increases in aldosterone. Similarly, in a cardiorenal substudy of EPHESUS, therapy with eplerenone led to a significant increase in aldosterone. These results confirm the blockade of mineralocorticoid receptors in these populations.
In dose ranging studies of chronic heart failure (NYHA classification II-IV), the addition of eplerenone to standard therapy resulted in expected dose dependent increases in aldosterone. Similarly, in a cardiorenal substudy of EPHESUS, therapy with eplerenone led to a significant increase in aldosterone. These results confirm the blockade of mineralocorticoid receptors in these populations.

 Patients with stage 1 and 2 CKD were started on eplerenone 25 mg or matching placebo daily. They could be uptitrated to eplerenone 50 mg daily, or matching placebo, if serum potassium at 4 weeks was < 5.0 mmol/L. Subsequently, patients received a maintenance dose that ensured that serum potassium did not exceed 5.0 mmol/L. If serum potassium rose above 5.0 mmol/L, patients were down titrated to a daily dose of 25 mg or matching placebo. Similarly, patients taking 25 mg or matching placebo, daily, were down titrated to 25 mg or matching placebo, every other day, if serum potassium was > 5.0 mmol/L.
Patients with stage 1 and 2 CKD were started on eplerenone 25 mg or matching placebo daily. They could be uptitrated to eplerenone 50 mg daily, or matching placebo, if serum potassium at 4 weeks was < 5.0 mmol/L. Subsequently, patients received a maintenance dose that ensured that serum potassium did not exceed 5.0 mmol/L. If serum potassium rose above 5.0 mmol/L, patients were down titrated to a daily dose of 25 mg or matching placebo. Similarly, patients taking 25 mg or matching placebo, daily, were down titrated to 25 mg or matching placebo, every other day, if serum potassium was > 5.0 mmol/L.
