1 Name of Medicine
Elafibranor.
2 Qualitative and Quantitative Composition
Each Iqirvo film-coated tablet contains 80 mg of elafibranor as the active ingredient.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Iqirvo 80 mg film-coated tablets are round, orange, debossed with 'ELA 80' on one side.
4.1 Therapeutic Indications
Iqirvo is indicated for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults with an inadequate response to UDCA, or as monotherapy in adults unable to tolerate UDCA.
4.2 Dose and Method of Administration
Dosage.
The recommended dose is 80 mg once daily with or without food.
Missed doses.
If a dose of elafibranor is missed, the patient should not take the missed dose and instead take their subsequent dose at the next scheduled time point. The patient should not take a double dose to make up for the missed dose.
Special populations.
Elderly patients.
No dose adjustment is necessary in patients older than 65 years of age (see Section 5.2 Pharmacokinetic Properties).
Patients with renal impairment.
No dose adjustment is necessary in patients with renal impairment (see Section 5.2 Pharmacokinetic Properties).
Patients with hepatic impairment.
No dose adjustment is necessary in patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment.
The safety and efficacy of elafibranor have not been established in patients with PBC with severe hepatic impairment. Use in patients with severe hepatic impairment (Child-Pugh C) is not recommended (see Section 5.2 Pharmacokinetic Properties).
Use of Iqirvo is not recommended in patients who have or develop decompensated cirrhosis (e.g. ascites, variceal bleeding, hepatic encephalopathy).
Method of administration.
For oral use. Take one tablet once daily.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients (see Section 6.1 List of Excipients).
4.4 Special Warnings and Precautions for Use
Liver related events.
Increases in liver biochemical tests including transaminases and bilirubin increase have been reported in 3.7% of participants receiving elafibranor compared to 5.7% of participants receiving placebo.
Clinical and laboratory assessment of liver function should be done prior to initiation of elafibranor treatment and thereafter according to routine patient management.
If increases in liver biochemical tests and/or liver dysfunction are observed, prompt investigation of the cause is recommended and interruption of elafibranor treatment should be considered.
Myalgia, myopathy, and rhabdomyolysis.
Increases in blood creatine phosphokinase (CPK) have been reported in participants receiving elafibranor (3.7% in elafibranor group compared to 0% in placebo group). In addition to these reported CPK increases, there was one case of rhabdomyolysis which occurred in the pivotal phase 3 ELATIVE study in a participant with cirrhosis and ongoing treatment with an HMG-CoA reductase inhibitor. CPK should be evaluated prior to initiation of elafibranor treatment and thereafter according to routine patient management. Periodic CPK measurements may be considered in patients starting elafibranor treatment, especially those on concomitant HMG-CoA reductase inhibitors. Patients on elafibranor treatment should be advised to report any unexplained muscle symptoms such as pain, soreness, or weakness to their treating physician.
If increases in CPK or unexplained signs and symptoms of muscle injury are observed, prompt investigation of the cause is recommended and interruption of elafibranor treatment should be considered (see Section 4.8 Adverse Effects (Undesirable Effects)).
Embryo-foetal toxicity.
Based on data from animal studies, elafibranor may cause foetal harm when administered to a pregnant woman. Patients should be informed of the potential risk to the foetus if elafibranor is taken during pregnancy (see Section 4.6 Fertility, Pregnancy and Lactation).
The use of elafibranor is not recommended during pregnancy and in women of childbearing potential not using effective contraception (see Section 4.6 Fertility, Pregnancy and Lactation). The pregnancy status of women of childbearing potential should be checked prior to initiation of elafibranor treatment.
Women of childbearing potential should be advised to use effective contraception during treatment and for 3 weeks following the final dose of elafibranor.
Use in the elderly.
No data available.
Paediatric use.
No data available. There is no relevant use of elafibranor in the paediatric population (below 18 years of age) for the indication of PBC.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Based on in vitro and in vivo studies, no clinically relevant drug-drug interaction is expected by co-administering elafibranor with any other medicinal products.
Based on in vitro studies, CYP and UGT enzymes were shown not to play a major role in elafibranor metabolism. Drug-drug interactions (DDI) are expected to be limited with drugs that significantly alter CYP or UGT activity.
Clinical studies.
Warfarin (CYP2C9 substrate).
Concomitant administration of elafibranor with warfarin resulted in no increase in exposure (AUC, Cmax) of warfarin, and no difference in international normalized ratio (INR) compared to warfarin alone.
Simvastatin and atorvastatin (CYP3A, organic anion transporting polypeptides 1B1 (OATP1B1) and OATP1B3 substrates).
Concomitant administration of repeat doses of elafibranor with simvastatin, or atorvastatin, resulted in no increase in exposure (AUC, Cmax) of simvastatin or its β-Hydroxyacid metabolite, or atorvastatin.
Indomethacin (PTGR1 inhibitor).
Following clinical DDI studies, no effect on the clinical PK of elafibranor was observed with co-administration of indomethacin.
Sitagliptin (dipeptidyl peptidase-IV (DPP-IV) inhibitor).
No clinically significant effects were observed when co-administering elafibranor as a DDI perpetrator with sitagliptin.
In vitro studies.
Cytochrome P450 (CYP) inhibition and induction.
Elafibranor and GFT1007 were not considered inhibitors of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. No time-dependent CYP inhibition was observed by elafibranor and GFT1007.
Elafibranor and GFT1007 did not cause induction on CYP1A2, CYP2B6, and CYP3A4.
UGT inhibition.
Based on in vitro data elafibranor is not expected to inhibit UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B7, 2B10, and 2B15 at clinically relevant concentrations.
GFT1007 inhibited UGT1A6 but not UGT1A1, 1A3, 1A4, 1A9, 2B7, 2B10, and 2B15, however this is not expected to be clinically relevant.
Transporter systems.
Elafibranor was an inhibitor of organic anion transporting polypeptides 1B3 (OATP1B3) and breast cancer resistance protein (BCRP) but was not an inhibitor of Permeability-glycoprotein/multidrug resistance protein 1 (P-gp/MDR1), OATP1B1, organic cation transporter 1 (OCT1), OCT2, organic anion transporter 1 (OAT1), multidrug and toxin extrusion protein 1 (MATE1), MATE2-K, OAT3 and bile salt export pump (BSEP).
GFT1007 was not considered an inhibitor of OAT3, OATP1B3, BSEP, P-gp/MDR1, BCRP, OATP1B1, OCT1, OCT2, OAT1, MATE1 and MATE2-K.
GFT3351 is an inhibitor of multidrug resistance associated protein 2 (MRP2) and MRP3. The clinical significance of MRP2 and MRP3 inhibition by GFT3351 is unknown. GFT3351 did not inhibit BCRP, P-gp, OATP1B1, OATP1B3, OAT1, OAT2, OAT3, OCT1, OCT2, MATE1, MATE2-K, and BSEP.
Elafibranor was found in vitro to be a substrate for intestinal transporters multidrug resistance-associated protein 2 (MRP2) and BCRP. Elafibranor and GFT1007 are not substrates of P-gp, OATP1B1, OATP1B3, OAT1, OAT3 and OCT2. The role of active efflux transport is considered to be negligible compared to the passive, concentration-gradient driven, high permeability absorption of elafibranor.
GFT3282 was a substrate of MRP2 and MRP3.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No human data on the effect of elafibranor on fertility are available.
In the first fertility study, following repeated oral administration in male (from 29 days prior to mating until sacrifice, i.e. for 7 weeks in total) and female (15 days prior to pairing until day 7 post-coitum) rats, no effects on oestrous cycling, mating indices, mating period and on absolute and relative weights of testes, epididymides or gravid uterus up to 100 mg/kg/day (estimated 17- and 14-times the combined AUC for elafibranor and GFT1007 estimated exposures at the Maximum Human Recommended Dose (MHRD), in male and female rats, respectively). There were no changes in sperm parameters up to 30 mg/kg/day (estimated 6.5-times the combined AUC for elafibranor and GFT1007 estimated exposures at the MHRD). In a second fertility and early embryonic development study, following repeated oral administration in male rats (from 4 weeks prior to mating until sacrifice, i.e. for 9 weeks in total), no effect on male fertility were observed up to 100 mg/kg/day (estimated 17 times the combined AUC for elafibranor and GFT1007 estimated exposures at the MHRD).
(Category D)
There is limited amount of data from the use of elafibranor in pregnant women.
Studies in pregnant animals with elafibranor indicate adverse effects (fetal loss, malformations, stillbirths and/or perinatal deaths) at clinically relevant exposure.
Elafibranor is not recommended during pregnancy because of potential harm to the fetus.
Elafibranor is not recommended in women of childbearing potential not using effective contraception because of potential harm to the fetus.
Women of childbearing potential should continue to use effective contraception for 3 weeks following the final dose of elafibranor. The pregnancy status of patients of childbearing potential should be checked prior to initiation of elafibranor treatment.
Women planning to become pregnant are recommended to consult with their physician regarding alternate treatment options (see Section 4.4 Special Warnings and Precautions for Use).
In pregnant rats, once daily oral administration of elafibranor during the period of organogenesis resulted in no effect on embryofetal development at dose up to 300 mg/kg/day (~100-fold the combined estimated AUC for elafibranor and GFT1007 exposures at the MHRD).
In pregnant rabbits, once daily administration of elafibranor during the period of organogenesis at the dose of 300 mg/kg/day (3.1-fold the combined estimated AUC for elafibranor and GFT1007 exposures at MHRD), was associated with marked maternal toxicity, increased embryo-lethality, reduced fetal weight plus a low incidence of fetal malformations. At 100 mg/kg/day (0.5-fold AUC exposure at the MHRD), despite maternal toxicity, there was no effect on embryofetal survival, or fetal weight nor fetal malformations, however, skeletal variations involving delayed and/or incomplete ossification of distal limb bones were observed. At 300 mg/kg/day (3.1-fold the combined estimated AUC for elafibranor and GFT1007 exposures at the MHRD), skeletal variations included delayed/incomplete ossification of multiple bones (metacarpal, median phalanx of hindpaw and talus), multiple visceral (absence of innominate artery) and external variations (malrotated paw, shortened tail) and malformations (cardiomegaly, absent kidney and ureter, fused/misaligned caudal vertebrae) were observed. No adverse effects were seen on embryofetal development at the low dose of 30 mg/kg/day (0.1-fold the combined estimated AUC for elafibranor and GFT1007 exposures at the MHRD). NOAEL for embryofetal toxicity was 30 mg/kg/day.
In the rat pre- and post-natal development (PPND) study, once daily oral administration of elafibranor during organogenesis through pregnancy and lactation was associated with reduced pup survival (during postnatal days 1-4 at doses 17-fold above combined maternal AUC for elafibranor and GFT1007 exposure at MHRD) and postnatal days 5-21 at 10 and 30 mg/kg/day (2.1- and 4.5-fold the combined maternal AUC for elafibranor and GFT1007 exposures at the MHRD, respectively), blue/black discolouration of the caudal section of some pups, lower pup body weights, and developmental delays (pinna unfolding) at all doses (2.1-fold the combined maternal AUC for elafibranor and GFT1007 exposures at the MHRD). Developmental delays were likely caused by the decrease in body weight. Adverse effects in the offspring occurred at maternal exposures at or above 2.5-times the combined AUC for elafibranor and GFT1007 exposures at MHRD. Increase in stillbirths were observed at 100 mg/kg/day (17-times the combined maternal AUC for elafibranor and GFT1007 exposures at MHRD). Stillbirths at 100 mg/kg/day and a single stillborn at 30 mg/kg/day (4.5-times the combined maternal AUC for elafibranor and GFT1007 exposures at MHRD) showed evidence of aortic or iliac arterial thrombosis (generally accompanied by vessel dilation, with minimal associated inflammation of the vessel wall, surrounding connective tissue, or adjacent abdominal structures). In pups sampled on postnatal day 14, elafibranor was not detected and only minimal plasma exposure to its active metabolite was detectable in pups from 100 mg/kg/day (maternal exposures were 17-fold above combined AUC for elafibranor and GFT1007 exposures at MHRD). The surviving adult offspring showed no effects of elafibranor on learning and memory, reflex development, or reproductive capability.
It is unknown whether elafibranor or its metabolites are excreted in human milk. In PPND study in rats, following oral administration of elafibranor to female rats through pregnancy and lactation, there were reduced survival and growth of offspring from 10 mg/kg/day (maternal exposures 2.1-times the combined AUC for elafibranor and GFT1007 at the MHRD). Low levels of GFT1007 were detected in 2 pups at 100 mg/kg/day (maternal exposures 17 times the combined AUC for elafibranor and GFT1007 at the MHRD), however, it is unclear whether excretion of elafibranor or its metabolite in milk contributed to the adverse effects on offspring.
Elafibranor is not recommended during breastfeeding and for at least 3 weeks following last dose of elafibranor because the risk to breastfed child cannot be excluded.4.7 Effects on Ability to Drive and Use Machines
Elafibranor has no influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
Summary of safety profile.
In the pivotal phase 3 ELATIVE study, 161 participants were randomized in a 2:1 ratio to receive elafibranor 80 mg (n=108) or placebo (n=53) for at least 52 weeks. At the end of the double-blind (DB) period of the study, the median duration of exposure was 63.07 and 61.00 weeks in the elafibranor and placebo groups, respectively.
The most commonly reported adverse drug reactions associated with elafibranor in more than 10% of participants (n=108) were abdominal pain (11.1%), diarrhoea (11.1%), nausea (11.1%) and vomiting (11.1%). These were non-serious and mild to moderate in severity.
The most common adverse drug reaction leading to treatment discontinuation was blood CPK increased (3.7%).
The most common adverse events with Iqirvo in the clinical study (reported in ≥ 5% and higher compared to placebo) were weight gain, arthralgia, constipation, muscle injury, fracture, gastroesophageal reflux disease, dry mouth, weight loss, and rash.
Tabulated list of adverse reactions.
Within the system organ class, the adverse reactions are listed by frequency using the following categories: very common (≥ 1/10), common (≥ 1/100 to < 1/100), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1000), very rare (< 1/10,000), not known (cannot be estimated from the available data). See Table 1.
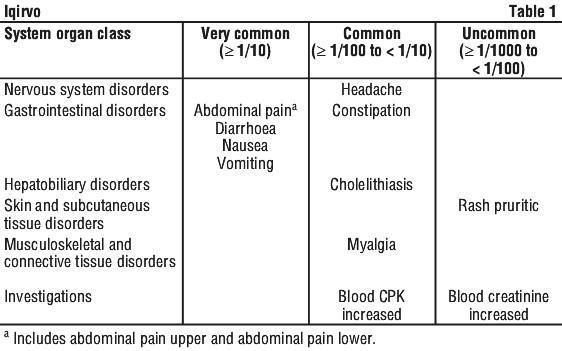
Description of selected adverse reactions.
Nine (8.3%) participants in the elafibranor group and 6 (11.3%) participants in the placebo group experienced headache. However, within the first 10 days of study treatment, more participants in the elafibranor group experienced headache compared to the placebo group (3.7% compared to 0% respectively).
Four (3.7%) participants in the elafibranor group and no participants in the placebo group had clinically significant blood CPK increase, leading to drug discontinuation. In 2 of the 4 participants, the CPK was > 5 x upper limit of normal (ULN). All events were non-serious and mild to moderate in intensity. Two of the participants also experienced associated symptom of myalgia. At baseline, mean CPK values were similar between the treatment groups and within normal range; values at Week 52 remained within normal range in both groups. The mean change from baseline at Week 52 was 6.2 (38.1) U/L in the elafibranor group and 12.3 (67.0) U/L in the placebo group.4.9 Overdose
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Bile and liver therapy, other drugs for bile therapy.
ATC code: A05AX06.
Mechanism of action.
Elafibranor and its main active metabolite GFT1007 are peroxisome proliferator-activated receptor (PPAR) agonists, both of which activate PPARα, PPARγ, and PPAR-δ in vitro. In vitro, both elafibranor and GFT1007 demonstrated 3- to 8-fold higher activity for PPAR-α compared to PPAR-γ and PPAR-δ. Although the in vitro pharmacology studies detected PPAR-γ activation by elafibranor and its metabolite GFT1007, toxicology studies in rats and monkeys (species with plasma metabolite profiles comparable to human) showed none of the adverse effects that are associated with PPAR-γ activation.
PPARα/δ are thought to be key regulators of bile acid (BA) homeostasis, inflammation and fibrosis. Activation of PPARα decreases bile acid (BA) synthesis, increases BA detoxification, and modulates BA output, resulting in decreased bile toxicity, and less injury to cholangiocytes and hepatocytes.
Activation of PPAR-δ also regulates transporters that absorb and secrete bile components, contributing this way to decreased bile toxicity and improving cholestasis.
Activation of PPARα and PPAR-δ also has anti-inflammatory effects by acting on different pathways of inflammation, nuclear factor kappa B (NF-κB) and B-cell lymphoma 6 (BCL6) pathways, respectively.
Pharmacodynamic effects.
In the pivotal phase 3 ELATIVE study, treatment with elafibranor resulted in a marked reduction from baseline in alkaline phosphatase (ALP) as early as 4 weeks which was sustained through week 52. In alignment with the observed biochemical response, greater reductions in biomarkers of BA synthesis including the BA precursor 7 alpha-hydroxy-4-cholesten-3-one (C4) and Fibroblast Growth Factor-19 (FGF-19), a BA synthesis regulator, were observed with elafibranor treatment. Significant decreases in Immunoglobulin M (IgM), Immunoglobulin G (IgG), and anti-inflammatory markers, were observed in participants treated with elafibranor compared to placebo in alignment with the in vitro demonstration of anti-inflammatory properties of elafibranor.
In vitro studies in human macrophages, monocytes and endothelial cells showed the capacity of elafibranor and/or GFT1007 to decrease the secretion of inflammatory markers such as Monocyte Chemoattractant Protein-1 (MCP-1) and Interleukin-6 (IL-6) through combined PPARα and PPAR-δ activation and parallel PPAR-independent mechanisms.
Anti-fibrotic properties of elafibranor were demonstrated in human primary hepatic stellate cells (hHSCs), pivotal for fibrogenesis in the liver. Elafibranor inhibits Platelet-Derived Growth Factor (PDGF)-stimulated hHSC proliferation in a dose-dependent manner via modulation of PDGFRβ phosphorylation. Additionally, elafibranor inhibits Transforming Growth Factor Beta (TGFβ1)-induced hHSC activation at the gene level, by down-regulating, in a dose-dependent manner, the expression of several fibrosis markers, such as alpha Smooth Muscle Actin (αSMA), Collagen 1 alpha 1 (Col1α1) and Collagen 4 alpha 1 (Col4α1), but without inhibiting the kinase activity of the TGFβ1 receptors.
Cardiac electrophysiology.
Thorough QT (TQT) analysis excluded any prolongation effect of elafibranor on QT/QTc interval at repeat doses of up to 300 mg for 14 days.
In clinical studies, no clinically meaningful changes in vital signs or in electrocardiogram (ECG) (including QTc interval) were observed in participants treated with elafibranor.
Clinical trials.
The efficacy of elafibranor was evaluated in Study GFT505B-319-1 (ELATIVE), a phase 3, randomised, DB, placebo-controlled study followed by an open-label long-term extension (OLE) in 161 adults with PBC with an inadequate response or intolerance to UDCA. Participants were randomized in a 2:1 ratio to receive elafibranor 80 mg or placebo once daily for at least 52 weeks. When applicable, participants continued their pre-study dose of UDCA throughout the study. Participants were included in the study if their ALP was ≥ 1.67 x ULN and total bilirubin (TB) was ≤ 2 x ULN. Participants were excluded in case of decompensated cirrhosis or other causes of liver disease.
Overall, the mean age was 57.1 years, and the mean weight was 70.8 kg. The study population was predominately female (96%) and white (91%). The baseline mean ALP concentration was 321.9 U/L and 39% of participants had a baseline ALP concentration > 3 x ULN. Mean baseline AST and ALT was 45.7 U/L and 49.6 U/L, respectively.
The mean baseline TB concentration was 9.6 micromol/L and 96% of participants had a baseline TB concentration ≤ ULN. The mean baseline liver stiffness measurement (LSM) by transient elastography was 10.1 kPa. The baseline mean PBC Worst Itch Numeric Rating Scale (NRS) score was 3.3 and 41% had moderate-to-severe pruritus at baseline (PBC Worst Itch NRS score ≥ 4); for those with moderate-to-severe pruritus, the baseline mean PBC Worst Itch NRS score was 6.2 for participants in the elafibranor 80 mg group and 6.3 for participants in the placebo group. The majority (n=153, 95%) of participants received treatment in combination with UDCA or as monotherapy in 5% (n=8) of participants who were unable to tolerate UDCA.
The primary endpoint was cholestasis response at week 52 as defined as the composite endpoint: ALP < 1.67 x ULN and TB ≤ ULN and ALP decrease ≥ 15%. The key secondary endpoints were ALP normalization at week 52 and the change in pruritus from baseline through week 52 and through week 24 based on the PBC Worst Itch NRS score in participants with moderate-to-severe pruritus at baseline.
Table 2 shows the primary composite endpoint of cholestasis response and the key secondary endpoint of ALP normalization.
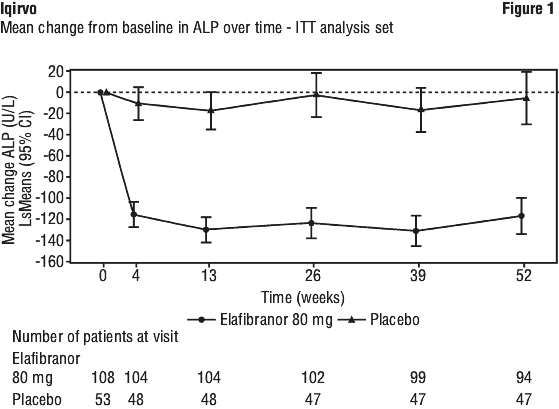
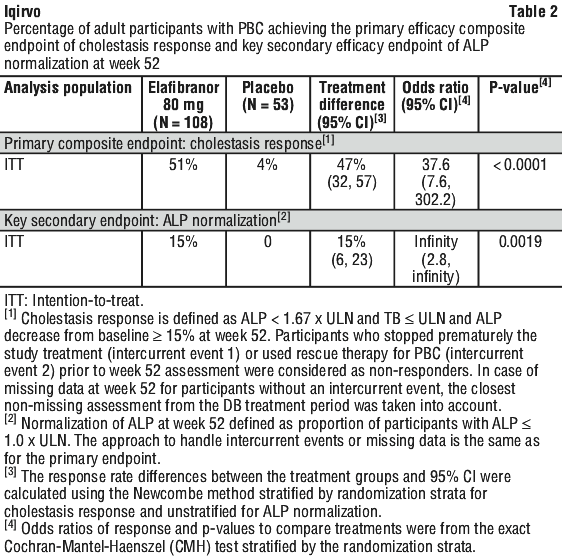 A significant decrease from baseline was seen as early as week 4 and was sustained over 52 weeks of treatment in the elafibranor group compared to placebo (Figure 1).
A significant decrease from baseline was seen as early as week 4 and was sustained over 52 weeks of treatment in the elafibranor group compared to placebo (Figure 1).
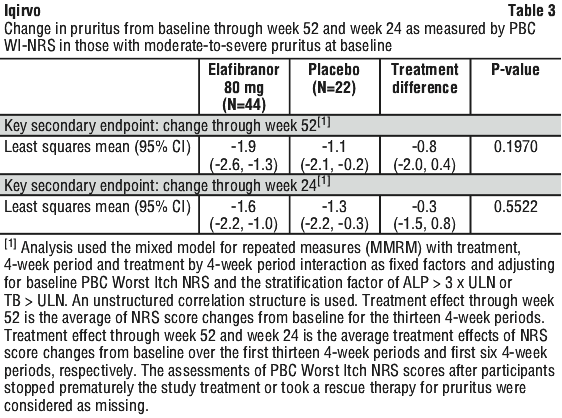
Patient-reported outcomes.
In participants with moderate-to-severe pruritus at baseline, the mean change from baseline in PBC Worst Itch NRS score through Week 52 and Week 24 decreased in participants randomized to elafibranor compared to placebo but did not reach statistical significance (Table 3).
 Treatment with elafibranor was associated with an improvement in pruritus as evidenced by a reduction in the PBC-40 Itch and 5-D Itch total scores compared to placebo at Week 52 (Table 4).
Treatment with elafibranor was associated with an improvement in pruritus as evidenced by a reduction in the PBC-40 Itch and 5-D Itch total scores compared to placebo at Week 52 (Table 4).

Lipid parameters.
Elafibranor demonstrated a favourable effect on lipid parameters. The mean reduction in very low-density lipoprotein-cholesterol (VLDL-C) and triglycerides (TG) was greater in participants treated with elafibranor compared to placebo at Week 52. The LSM means difference from placebo in VLDL-C was -0.1 mmol/L [(95% CI: -0.2, -0.1); p < 0.001] and for TG was -0.3 mmol/L [(95% CI: -0.4, -0.1)]; p < 0.001]. High-density lipoprotein-cholesterol (HDL-C) remained stable on treatment with elafibranor.
Mean reduction in IgM.
The mean reduction in IgM was greater in participants treated with elafibranor compared to placebo at Week 52. The LSM means difference from placebo in IgM was -0.6 g/L [(95% CI: -0.9, -0.3); p < 0.001].
5.2 Pharmacokinetic Properties
Absorption.
Elafibranor plasma exposure (AUC) increases proportionally from 50 to 360 mg (0.6 to 4.5 times the recommended dosage). Steady-state is achieved by day 14 following once daily dosing. The pharmacokinetics (PK) of elafibranor and its major active metabolite GFT1007 was found to be time-independent after 16-day repeated administration. Elafibranor and its active metabolite exposure in participants with PBC are listed in Table 5.
 Following repeated oral administration in participants with PBC, median peak plasma levels of elafibranor and GFT1007 at doses of 80 mg occur within 1.25 hours.
Following repeated oral administration in participants with PBC, median peak plasma levels of elafibranor and GFT1007 at doses of 80 mg occur within 1.25 hours.
When administered with a high-fat and high-calorie meal, there was a 30-minute delay in Tmax for elafibranor and a 1 hour delay for GFT1007 in fed compared to fasted conditions. The plasma Cmax and AUC of elafibranor decreased by 50% and 15% respectively and the plasma Cmax of GFT1007 decreased by 30% but the AUC was not affected. Given the 2.2-5.3-fold higher circulating plasma levels of the pharmacologically active metabolite GFT1007 compared to elafibranor, food intake was deemed to have limited clinical impact based on overall exposure of parent and active metabolite.
Distribution.
Plasma protein binding of both elafibranor and GFT1007 is approximately 99.7% (mainly to serum albumin). The mean apparent volume of distribution (Vd/F) of elafibranor in humans is 4731 L, following single dose of elafibranor at 80 mg in fasted conditions.
Metabolism.
In vitro, elafibranor is metabolised by 15-ketoprostaglandin 13-Δ reductase (PTGR1) to form metabolite GFT1007. Other enzymes involved in the metabolism of elafibranor also included CYP2J2, and uridine diphosphate (UDP)-glucuronosyltransferase (UGT) isoforms, UGT1A3, UGT1A4, and UGT2B7. GFT1007 was further metabolised by CYP2C8 and UGT1A3 and UGT2B7.
Following oral administration of 14C radiolabelled elafibranor, it was rapidly hydrolysed to the active metabolite GFT1007. Two major metabolites were identified in plasma, GFT1007 (active metabolite) and glucuronide conjugates (inactive metabolites).
Excretion.
Following single 80 mg dose under fasted conditions, mean elimination half-life is 68.2 hours for elafibranor, and 15.4 hours for metabolite GFT1007. Elafibranor mean apparent total clearance (CL/F) was 50.0 L/h after a single 80 mg dose under fasted conditions.
Following a single 120 mg oral dose of 14C radiolabelled elafibranor in healthy participants, approximately 77.1% of the dose was recovered in faeces, primarily as elafibranor (56.7% of the administered dose) and its active metabolite GFT1007 (6.08% of the administered dose). Approximately 19.3% recovered in urine, primarily as glucuronide conjugates.
Pharmacokinetics in special patient populations.
There was no evidence that age (from 18 to 80 years old), gender, race, Body Mass Index (BMI), and renal status, had any clinically meaningful impact on elafibranor and GFT1007 PK.
Hepatic impairment.
The total drug exposure of the parent and active metabolite was not significantly different between participants with normal hepatic function and hepatically impaired participants (Child Pugh A, B and C). No dose adjustment is required for patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment. However, the unbound fraction of elafibranor and GFT1007 increased by approximately 3-fold in the severe (Child Pugh C) hepatically impaired participants. Elafibranor is not recommended for patients with severe hepatic impairment (Child-Pugh C).
5.3 Preclinical Safety Data
Genotoxicity.
Elafibranor was negative in the in vitro bacterial reverse mutation (Ames) test and the in vivo micronucleus test. A potential clastogenic activity was observed with elafibranor at the thymidine kinase locus in mouse lymphoma cells in the in vitro micronucleus test. The result was not confirmed in two in vivo micronucleus tests in the rat bone marrow. Additionally, no genotoxic activity of elafibranor on hepatocytes was observed in two in vivo genotoxicity studies in the rat liver (unscheduled DNA synthesis assay and Comet assay). Together, these tests demonstrated that elafibranor has no risk of mutagenicity or genotoxicity after oral administration.
The metabolites GFT1007 and racemic GFT3351 (acyl glucuronide metabolite) were both negative in the in vitro bacterial reverse mutation (Ames) assay. GFT1007 tested negative in the in vitro micronucleus test in L5178Y tk+/- mouse lymphoma cells, and GFT3351 tested negative in the in vitro micronucleus assay in human lymphocytes.
Carcinogenicity.
Elafibranor was assessed in two carcinogenicity studies in mice and rats, with oral gavage administration for up to two years at 1, 3, 10, or 30 mg/kg/day.
In the 2-year carcinogenicity study in mice, oral administration of elafibranor produced neoplastic findings limited to hepatocellular carcinoma, adenoma and adenoblastoma, in both sexes, present at subclinical exposures.
In the 2-year carcinogenicity study in rats, oral administration of elafibranor produced hepatocellular tumours (adenoma or carcinoma) in males and females at clinically relevant exposures. At 30 mg/kg/day (6-times the recommended dose based on combined AUC for elafibranor and GFT1007) in male rats, there was an increase in testicular Leydig cell adenomas and benign pancreatic acinar cell adenomas.
Treatment-related liver tumours identified in both the mouse and rat carcinogenicity studies may be attributed to the expected rodent-specific PPARα-related liver toxicity and its related consequences and are not relevant to humans. Thus, up to the highest tested dose in both species (i.e. 30 mg/kg/day, approximately 3- and 6-fold the combined AUC exposure for elafibranor and GFT1007 at the maximum human recommended dose (MHRD) of 80 mg/day), elafibranor does not result in an increased incidence of tumours that are relevant to humans.6 Pharmaceutical Particulars
6.1 List of Excipients
Tablet content.
Microcrystalline cellulose; povidone; croscarmellose sodium; magnesium stearate; colloidal anhydrous silica.
Film-coating.
Polyvinyl alcohol; titanium dioxide; macrogol 3350; purified talc; iron oxide yellow; iron oxide red.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Iqirvo film-coated tablets are packaged in a HDPE bottle with a child-resistant polypropylene closure with integrated desiccant unit.
Each pack contains 30 film-coated tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Elafibranor is a yellow crystalline powder. It is practically insoluble in aqueous buffer and water, sparingly soluble in methanol, and freely soluble in organic solvents.
Chemical structure.
The molecular formula of elafibranor is C22H24O4S.
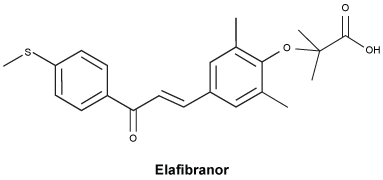 IUPAC name: 2-[2,6-dimethyl-4-[(E)-3-(4-methylsulfanylphenyl)-3-oxoprop-enyl]phenoxy]-2-methylpropanoic acid.
IUPAC name: 2-[2,6-dimethyl-4-[(E)-3-(4-methylsulfanylphenyl)-3-oxoprop-enyl]phenoxy]-2-methylpropanoic acid.
Molecular weight: 384.49 g/mol.
CAS number.
923978-27-2.7 Medicine Schedule (Poisons Standard)
S4.