1 Name of Medicine
Osilodrostat phosphate.
2 Qualitative and Quantitative Composition
Isturisa 1 mg.
Each film-coated tablet contains osilodrostat phosphate corresponding to 1 mg osilodrostat.
Isturisa 5 mg.
Each film-coated tablet contains osilodrostat phosphate corresponding to 5 mg osilodrostat.
Isturisa 10 mg.
Each film-coated tablet contains osilodrostat phosphate corresponding to 10 mg osilodrostat.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Film-coated tablet.
Isturisa 1 mg film-coated tablets.
Pale yellow, round, biconvex bevelled-edge tablets, unscored, debossed with '1' on one side. Approximate diameter 6.1 mm.
Isturisa 5 mg film-coated tablets.
Yellow, round, biconvex bevelled-edge tablets, unscored, debossed with '5' on one side. Approximate diameter 7.1 mm.
Isturisa 10 mg film-coated tablets.
Pale orange brown, round, biconvex bevelled-edge tablets, unscored, debossed with '10' on one side. Approximate diameter 9.1 mm.4.1 Therapeutic Indications
Isturisa is indicated for the treatment of endogenous Cushing's syndrome in adults.
4.2 Dose and Method of Administration
Treatment should be initiated and supervised by physicians experienced in endocrinology or internal medicine and with access to the appropriate facilities for monitoring of biochemical responses since the dose must be adjusted to meet the patient's therapeutic needs, based on the normalisation of cortisol levels.
Dose.
The recommended starting dose is 2 mg osilodrostat twice daily. For patients of Asian ancestry, a reduced starting dose of 1 mg twice daily is recommended (see Section 5.2 Pharmacokinetic Properties).
The dose can be gradually titrated (initially by dose increments of 1 or 2 mg) based on individual response and tolerability, with the aim to achieve normal cortisol levels. It is recommended that cortisol levels (e.g. 24-hour urinary free cortisol, serum/plasma cortisol) be monitored every 1-2 weeks until adequate clinical response is maintained. Thereafter, cortisol levels should be monitored at regular intervals, and reasons for additional monitoring should be considered (see Section 4.4; Section 4.5). Increases in dose should not occur more frequently than once every 1-2 weeks and should be guided by the results of cortisol assessments and by the individual clinical response.
The dose of osilodrostat should be decreased or treatment temporarily interrupted if cortisol levels are below the lower limit of normal, or if there is a rapid decrease in cortisol levels to the lower part of the normal range, or if the patient has signs or symptoms suggestive of hypocortisolism (see Section 4.4 Special Warnings and Precautions for Use). Isturisa may be resumed after resolution of symptoms at a lower dose, provided that cortisol levels are above the lower limit of normal in the absence of glucocorticoid substitution. Management of other suspected adverse reactions at any time during treatment may also require a temporary dose reduction or temporary interruption of treatment.
The usual maintenance dose in clinical studies varied between 2 and 7 mg twice daily. The maximum recommended dose of Isturisa is 30 mg twice daily.
If a dose is missed, the patient should take the prescribed dose at the next scheduled time; the next dose should not be doubled.
Elderly (65 years or above).
There is no evidence to suggest that dose adjustment is required in patients aged 65 years or above. However, data on the use of osilodrostat in this population are limited and Isturisa should therefore be used with caution in this age group.
Renal impairment.
No dose adjustment is required for patients with renal impairment (see Section 5.2 Pharmacokinetic Properties). Urinary free cortisol (UFC) levels should be interpreted with caution in patients with moderate to severe renal impairment, due to reduced UFC excretion. Alternative methods for cortisol monitoring should be considered in these patients.
Hepatic impairment.
No dose adjustment is required for patients with mild hepatic impairment (Child-Pugh A). For patients with moderate hepatic impairment (Child-Pugh B), the recommended starting dose is 1 mg twice daily. For patients with severe hepatic impairment (Child-Pugh C), the recommended starting dose is 1 mg once daily in the evening, with initial up-titration to 1 mg twice daily (see Section 5.2 Pharmacokinetic Properties).
Data on use in patients with hepatic impairment is limited. More frequent monitoring of adrenal function may be required in patients with hepatic impairment during dose titration.
Paediatric population.
The safety and efficacy of Isturisa in patients less than 18 years of age have not yet been established. No data is available.
Method of administration.
Oral use.
Isturisa can be taken with or without food.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Hypocortisolism.
Patients should be monitored closely for hypocortisolism and potentially life-threatening adrenal insufficiency.
Inhibition of cortisol synthesis by osilodrostat has led to hypocortisolism-related events such as cortisol withdrawal syndrome (symptomatic decrease of cortisol levels, but still above the lower limit of the normal range) and adrenal insufficiency (cortisol levels below the normal range).
Cortisol levels should be monitored at regular intervals (see Section 4.2 Dose and Method of Administration), since hypocortisolism-related events can occur at any time during treatment and after treatment discontinuation, irrespective of dose. Additional monitoring is recommended especially during conditions of increased cortisol demand, such as physical or psychological stress, or during changes in concomitant medications that may affect osilodrostat exposure (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). It is recommended to use laboratory methods that do not exhibit significant cross-reactivity with cortisol precursors such as 11-deoxycortisol that may increase during osilodrostat treatment.
Patients should be alerted to the signs and symptoms associated with hypocortisolism (e.g. nausea, vomiting, fatigue, abdominal pain, loss of appetite and dizziness).
Symptomatic patients should be monitored for hypotension, hyponatraemia, hyperkalaemia and/or hypoglycaemia. If hypocortisolism is suspected, cortisol levels should be measured and temporary dose reduction or interruption of osilodrostat considered. After osilodrostat discontinuation, cortisol suppression may persist for months or longer, irrespective of osilodrostat administered dose or baseline disease severity. Patients should be closely and regularly monitored after interruption or discontinuation.
If necessary, corticosteroid substitution should be initiated. Isturisa may be resumed after resolution of symptoms at a lower dose, provided that cortisol levels are above the lower limit of normal in the absence of glucocorticoid substitution.
QTc prolongation.
In a thorough QT study, osilodrostat was associated with a dose-dependent QT interval prolongation (mean maximum estimated QTcF increase by +5.3 ms at the highest recommended dose of 30 mg) which may cause cardiac arrhythmias (see Section 5.1 Pharmacodynamic Properties). Adverse reactions of QT prolongation and clinically relevant ECG findings have been reported in clinical studies.
An ECG should be performed prior to the start of Isturisa treatment, within one week after treatment initiation, and as clinically indicated thereafter. If the QTc interval exceeds 480 ms prior to or during treatment, cardiology consultation is recommended. Temporary dose reduction or interruption may be required.
Any hypokalaemia, hypocalcaemia or hypomagnesaemia should be corrected prior to Isturisa administration and electrolyte levels should be monitored periodically during therapy.
Isturisa should be used with caution and the benefit-risk carefully weighed in patients with risk factors for QT prolongation such as:
congenital long QT syndrome;
significant cardiovascular disease (including congestive heart failure, recent myocardial infarction, unstable angina, sustained ventricular tachycardia, advanced heart block and clinically significant bradyarrhythmias); and
concomitant medicinal products known to prolong the QT interval (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
If Isturisa is used in patients with these risk factors, more frequent ECG monitoring is recommended.
Corticotroph tumour growth.
Discontinuation of osilodrostat treatment should be considered in patients who develop MRI verified corticotroph tumour invasiveness during treatment.
Concomitant use with strong enzyme inhibitors and inducers.
Caution and closer monitoring are advised when co-administered medicinal products that strongly inhibit or induce multiple enzymes are introduced or discontinued during osilodrostat treatment (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions), as they may affect osilodrostat exposure and may result in a risk of adverse events (due to a potential increase in exposure) or of decreased efficacy (due to a potential decrease in exposure).
Women of childbearing potential.
Isturisa may cause foetal harm. Pregnancy status should be verified in women of childbearing potential prior to the initiation of Isturisa, and these patients should be advised of a potential risk to the foetus and of the need to use effective contraception during treatment and for at least one week after stopping treatment (see Section 4.6 Fertility, Pregnancy and Lactation).
Elevations in adrenal hormone precursors and androgens.
Osilodrostat blocks cortisol synthesis may increase circulating levels of adrenal hormone precursors and testosterone. Hypokalaemia should be corrected prior to initiating osilodrostat. Monitor patients treated with osilodrostat for hypokalaemia, worsening of hypertension and oedema. Additional therapy, dose reduction or interruption may be required. Increases in testosterone may lead to hirsutism and acne (in females). See Section 4.8 Adverse Effects (Undesirable Effects).
Use in the elderly.
Data on the use of osilodrostat in this population are limited and Isturisa should therefore be used with caution in this age group.
Paediatric use.
The safety and efficacy of Isturisa in patients less than 18 years of age have not yet been established. No data are available.
Effects on laboratory tests.
Blood corticotrophin increased and electrocardiogram QT prolonged (see discussion above). Blood testosterone and transaminases may be increased.4.5 Interactions with Other Medicines and Other Forms of Interactions
Potential pharmacodynamic interactions.
Co-administration of osilodrostat with other therapies known to affect the QT interval can lead to QT prolongation in patients with known cardiac rhythm disorders (see Section 4.4 Special Warnings and Precautions for Use; Section 5.1 Pharmacodynamic Properties). A washout period should be considered when switching from other products known to affect the QT interval such as pasireotide or ketoconazole.
Effects of other medicinal products on the pharmacokinetics of osilodrostat.
The potential for clinical drug-drug interactions (DDI) with concomitantly administered medicinal products that inhibit transporters or a single CYP or UGT enzyme is low (see Section 5.2 Pharmacokinetic Properties).
Based on in vitro studies, osilodrostat is not a substrate for OATP1B1 or OATP1B3 transporters.
Strong enzyme inhibitors.
Caution is advised when co-administered medicinal products that strongly inhibit multiple enzymes are introduced or discontinued during osilodrostat treatment (see Section 4.4 Special Warnings and Precautions for Use).
Strong enzyme inducers.
Caution is advised when co-administered medicinal products that strongly induce multiple enzymes (e.g. rifampin) are introduced or discontinued during osilodrostat treatment (see Section 4.4 Special Warnings and Precautions for Use).
Effects of osilodrostat on the pharmacokinetics of other medicinal products.
Because osilodrostat and its major metabolite M34.5 may inhibit and/or induce multiple enzymes and transporters, general caution is advised when osilodrostat is co-administered with sensitive enzyme or transporter substrates with a narrow therapeutic index. Available interaction data is summarised below (also see Section 5.2 Pharmacokinetic Properties).
Clinical studies. In a healthy volunteer study (n=20) using a single dose of 50 mg osilodrostat and a probe drug cocktail, osilodrostat was found to be a mild inhibitor of CYP2D6 and CYP3A4/5, a mild to moderate inhibitor of CYP2C19, and a moderate inhibitor of CYP1A2.
CYP2D6.
AUC geometric mean ratio of 1.5 for dextromethorphan (CYP2D6 substrate) when dosed with osilodrostat compared to when dosed alone.
CYP3A4.
AUC geometric mean ratio of 1.5 for midazolam (CYP3A4 substrate) when dosed with osilodrostat compared to when dosed alone.
CYP2C19.
AUC geometric mean ratio of 1.9 for omeprazole (CYP2C19 substrate) when dosed with osilodrostat compared to when dosed alone. However, an in vitro signal of time-dependent inhibition has been observed, thus the consequence following repeated dosing is unclear. Osilodrostat should be used with caution when coadministered with sensitive CYP2C19 substrates with a narrow therapeutic index.
CYP1A2.
AUC geometric mean ratio of 2.5 for caffeine (CYP1A2 substrate) when dosed with osilodrostat compared to when dosed alone. However, an in vitro signal of CYP1A2 induction has been observed, thus the consequence following repeated dosing is unclear. Osilodrostat should be used with caution when co-administered with sensitive CYP1A2 substrates with a narrow therapeutic index such as theophylline and tizanidine.
In a healthy volunteer study (n=24), osilodrostat (30 mg twice daily for 7 days before concomitant administration with a combined oral contraceptive containing 0.03 mg ethinyl oestradiol and 0.15 mg levonorgestrel and continued for another 5 days) did not have a clinically meaningful effect on the AUC and Cmax of ethinyl estradiol (geometric mean ratio: 1.03 and 0.88, respectively) and AUC of levonorgestrel (geometric mean ratio: 1.02). The Cmax of levonorgestrel fell slightly outside the bioequivalence acceptance range (geometric mean ratio: 0.86; 90% confidence interval: 0.737-1.00). The effects of a longer induction period and an interaction with other hormonal contraceptives have not been studied (also see Section 4.4 Special Warnings and Precautions for Use; Section 4.6 Fertility, Pregnancy and Lactation).
In vitro data. In vitro data for osilodrostat and its major metabolite M34.5 suggest a potential for both inhibition and induction for CYP1A2, CYP2B6 and CYP3A4/5, a potential for time dependent inhibition of CYP2C19, and an inhibitory potential for CYP2D6, CYP2E1 and UGT1A1. It cannot be excluded that osilodrostat may affect the exposure of sensitive substrates for these enzymes.
In vitro data for osilodrostat and its major metabolite M34.5 suggest an inhibitory potential for OATP1B1, OCT1, OCT2, OAT1, OAT3 and MATE1. It cannot be excluded that osilodrostat may affect the exposure of sensitive substrates for these transporters.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There is no information on the effect of osilodrostat on human fertility.
In a fertility and early embryonic development study in rats oral maternal doses of 50 mg/kg (92-fold the exposure at the MRHD based on AUC) resulted in prolonged oestrous cycles and impaired fertility and embryo viability. The NOAEL was 5 mg/kg (8-fold the clinical AUC at the MRHD).
(Category D1)
There are no or limited amount of data from the use of osilodrostat in pregnant women. Studies in animals have shown reproductive toxicity.
Osilodrostat administration to pregnant rats during organogenesis at oral doses up to 5 mg/kg (9 times human dose based on AUC) did not adversely affect embryofetal development. Maternal toxicity, increased embryonic and fetal deaths, decreased fetal weights and malformations occurred at 50 mg/kg (86-times the maximum clinical dose based on AUC).
Osilodrostat administration to pregnant rabbits during organogenesis at oral doses up to 3 mg/kg (0.6 times human clinical exposure based on AUC) did not adversely affect embryofetal development. Maternal toxicity, increased embryo resorption decreased fetal viability was observed at 10 mg/kg (10 times the maximum clinical dose based on AUC). In a pre- and postnatal toxicity study in rats there were no adverse effects on growth, development or reproductive performance of offspring following maternal dosing at 5 mg/kg (10 times the maximum clinical dose based on AUC). Delayed parturition and dystocia and decreased pup survival were observed at 20 mg/kg (38 times the maximum clinical dose based on AUC).
Isturisa should not be used during pregnancy and in women of childbearing potential not using contraception.
It is unknown whether osilodrostat or its metabolites are excreted in human milk. A risk to the newborns/infants cannot be excluded. Breast-feeding should be discontinued during treatment with Isturisa and for at least one week after treatment.
Women of childbearing potential.
Based on preclinical data, osilodrostat may cause fetal harm when administered to a pregnant woman. A pregnancy test before initiating treatment is recommended in women of childbearing potential. Women of childbearing potential have to use effective contraception during and for at least one week after treatment. If hormonal contraceptives other than the oral combination of ethinylestradiol and levonorgestrel are used, an additional barrier method of contraception is recommended (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
1 Drugs which have caused, are suspected to have caused or may be expected to cause, an increased incidence of human fetal malformations or irreversible damage. These drugs may also have adverse pharmacological effects. Accompanying texts should be consulted for further details.4.7 Effects on Ability to Drive and Use Machines
Isturisa may have a minor influence on the ability to drive and use machines. Patients should be warned about the potential for dizziness and fatigue (see Section 4.8 Adverse Effects (Undesirable Effects)) and should be advised not to drive or use machines if these symptoms occur.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
A total of 210 patients with Cushing's disease has been treated with osilodrostat in the pivotal Phase III studies.
The most frequent (incidence ≥ 10%) adverse drug reactions (ADRs) reported in the pivotal Phase III studies (C2301 and C2302) with Isturisa were adrenal insufficiency (see Section 4.4 Special Warnings and Precautions for Use), fatigue, oedema, vomiting, nausea, decreased appetite, headache, dizziness, hypotension, arthralgia, myalgia, tachycardia and blood testosterone increased.
Also see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use.
Tabulated list of adverse drug reactions.
Adverse drug reactions (Table 1) are listed by MedDRA system organ class. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. Within each frequency grouping, adverse drug reactions are presented in order of decreasing seriousness. In addition, the corresponding frequency category for each adverse drug reaction is based on the following convention: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from the available data).

Description of selected adverse reactions.
CYP11B1 inhibition by osilodrostat is associated with adrenal steroid precursor accumulation and testosterone increases. In a clinical study with osilodrostat, mean testosterone levels in female patients increased from high normal at baseline to above the upper limit of the normal range. The increases reversed when treatment was interrupted. The testosterone increase was associated with mild to moderate cases of hirsutism or acne in a subset of patients.
ACTH values above 10-fold upper limit of normal were observed in some Cushing's disease patients treated with osilodrostat in the clinical studies (see Section 5.1 Pharmacodynamic Properties) and may be associated with cortisol values below the lower limit of normal.
Reporting of suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at https://www.tga.gov.au/reporting-problems.4.9 Overdose
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
Overdosage may result in severe hypocortisolism. Signs and symptoms suggestive of hypocortisolism may include nausea, vomiting, fatigue, low blood pressure, abdominal pain, loss of appetite, dizziness and syncope.
In case of suspected overdosage, Isturisa should be interrupted, cortisol levels checked, and if necessary, corticosteroid supplementation initiated. Close surveillance may be necessary including monitoring of the QT interval, blood pressure, glucose, fluid and electrolyte balance until the patient's condition is stable.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Anticorticosteroids, ATC code: H02CA02.
Mechanism of action.
Osilodrostat is a cortisol synthesis inhibitor. It inhibits 11β-hydroxylase (CYP11B1), the enzyme responsible for the final step of cortisol biosynthesis in the adrenal gland.
CYP11B1 inhibition is associated with the accumulation of precursors such as 11-deoxycortisol and acceleration of adrenal biosynthesis including androgens. In Cushing's disease, the fall in plasma cortisol concentration also stimulates ACTH secretion, via the feedback mechanism which accelerates steroid biosynthesis (see Section 4.8 Adverse Effects (Undesirable Effects)).
Pharmacodynamic effects.
In a thorough QT study (n=86 male and female healthy volunteers) with osilodrostat, the maximum QTcF interval duration differences to placebo were 1.73 ms (90% CI: 0.15, 3.31) at the 10 mg dose and 25.38 ms (90% CI: 23.53, 27.22) at a supratherapeutic dose of 150 mg. Based on an interpolation of these results, the mean maximum prolongation at the highest recommended dose of 30 mg is estimated to be +5.3 ms.
Clinical trials.
The efficacy and safety of osilodrostat in adult patients with Cushing's disease were evaluated in two phase III multicenter studies (study C2301 and C2302).
Study C2301 is a randomized withdrawal (RW) study, and Study C2302 is a double-blind, randomized study of osilodrostat vs placebo.
Study C2301. The study C2301 consisted of a 26-week open-label period of single-arm osilodrostat treatment period, followed by an 8-week double-blind randomised withdrawal period in which patients were randomised in 1:1 ratio to either osilodrostat or placebo and a subsequent osilodrostat 14-week open-label period. Patients who maintained clinical benefit on osilodrostat could continue in a long-term extension period until last patient achieved week 72, in order to collect further efficacy and safety data.
The eligibility criteria included Cushing's disease (with confirmation of the pituitary source of excess adrenocorticotrophic hormone), and a mean urinary free cortisol (mUFC, derived from three 24-hour urine collections) value greater than 1.5 times the upper limit of normal (ULN) at screening.
A total of 137 adult patients were enrolled. The mean age of the patients was 41.2 years, and the majority were female (77%). Seven patients were aged 65 years or older. Prior therapy included pituitary surgery in 88% of patients and prior medical therapy in 75% of patients. The mean and median baseline mUFC levels were 1006.0 nanomol/24 h (7 x ULN) and 476.4 nanomol/24 h (3 x ULN), respectively (ULN: 138 nanomol/24 h). Co-morbidities at baseline included hypertension (67.9% of patients), obesity (29.9%), diabetes mellitus (21.9%) and osteoporosis (27.7%).
Patients received a starting dose of 2 mg osilodrostat twice daily and the dose could be uptitrated based on individual response and tolerability during an initial 12-week period. Patients with no further dose increases during the following 12 weeks and with a mUFC ≤ ULN at week 24 were randomised in a 1:1 ratio at week 26 to receive either osilodrostat or matching placebo for 8 weeks (double-blind randomised withdrawal period), followed by open-label osilodrostat for the remainder of the study. At week 26, 71 patients were randomised in a 1:1 ratio to continue receiving osilodrostat (n=36) or to switch to placebo (n=35). Patients who were not eligible for randomisation at week 24 (n=47) continued on open-label osilodrostat treatment. 19 patients discontinued prior to week 26. 113 patients completed week 48 and 106 patients entered the extension phase. An additional 8 patients discontinued between week 48 and week 72.
The primary objective was to compare the proportion of complete responders at week 34 (the end of the 8-week randomised withdrawal period) between patients randomised to continued active treatment and placebo. For the primary endpoint, a complete response was defined as a mUFC value ≤ ULN at week 34. Patients whose dose was increased during the randomised withdrawal period or who discontinued randomised treatment were considered nonresponders. The key secondary endpoint was to assess the complete response rate at week 24. Patients with dose increases between weeks 12 and 24 and patients with no valid mUFC assessment at week 24 were counted as non-responders for the key secondary endpoint.
Results.
The study C2301 met its primary and key secondary endpoints (Table 2).
Median mUFC levels decreased to 62.5 nanomol/24 h (-84.1% change from baseline, n=125) at week 12, to 75.5 nanomol/24 h (-82.3%, n=125) at week 24 and to 63.3 nanomol/24 h (-87.9%, n=108) at week 48 and to 64 nanomol/24 h (-86.6%, n=96) at week 72.
 Improvements were observed in cardiovascular and metabolic parameters (Table 3) and 85.6% of patients with available assessments showed an improvement in at least one physical feature of Cushing's disease at week 48. With the longer follow up, improvements in cardiovascular and metabolic parameters and physical features of Cushing's disease were maintained.
Improvements were observed in cardiovascular and metabolic parameters (Table 3) and 85.6% of patients with available assessments showed an improvement in at least one physical feature of Cushing's disease at week 48. With the longer follow up, improvements in cardiovascular and metabolic parameters and physical features of Cushing's disease were maintained.
 Osilodrostat treatment also resulted in an improvement in patient-reported outcomes. Improvements from baseline above the established minimal important difference (MID) were observed for Cushing's QoL (total score, Physical Problems subscale and Psychosocial Issues subscale), EQ-5D Utility index and BDI-II (depression) scores. The mean Cushing QoL total score improved from 42.2 at baseline to 58.2 (+14.1; +52.4% change from baseline) at week 48. The improvements observed during the core phase were maintained during the extension phase.
Osilodrostat treatment also resulted in an improvement in patient-reported outcomes. Improvements from baseline above the established minimal important difference (MID) were observed for Cushing's QoL (total score, Physical Problems subscale and Psychosocial Issues subscale), EQ-5D Utility index and BDI-II (depression) scores. The mean Cushing QoL total score improved from 42.2 at baseline to 58.2 (+14.1; +52.4% change from baseline) at week 48. The improvements observed during the core phase were maintained during the extension phase.
Study C2302. Study C2302 recruited 74 adult patients with Cushing's disease. 73 patients received study treatment. The study comprised of a core phase of 12 weeks of a double-blind, placebo-controlled period, followed by a 36-week open-label treatment period with osilodrostat. The eligibility criteria included a mean urinary free cortisol value (mUFC, derived from three 24-hour urine collections) greater than 1.3 times the upper limit of normal (ULN=138 nanomol/24 h) at screening, and a confirmation of the pituitary source of excess Adrenocorticotropic Hormone (ACTH).
The mean age of the enrolled patients was 41.2 years, and 84% were female. Overall, 87.7% had undergone surgery prior to study entry and 12.3% of patients had received radiotherapy prior to study start. 54.2% and 76% of patients in the osilodrostat and placebo arms, respectively, had previously received medical treatment for Cushing's disease. Overall, the following relevant comorbidities were reported in the medical history of enrolled patients: hypertension (61.6%), obesity (13.7%), diabetes mellitus (16.4%), and osteoporosis (26.0%). The overall median and mean baseline mUFC levels were 340.3 nanomol/24 h (2.5 x ULN) and 431.7 nanomol/24 h (3 x ULN), respectively.
At baseline patients were randomly allocated in a 2:1 fashion to receive either osilodrostat 2 mg bid or matching-placebo; the dose could be gradually increased at 3-week intervals up to 20 mg bid. At the end of the 12-week double-blind randomised period, all patients were treated with open-label osilodrostat. The starting dose was 2 mg bid. Patients receiving daily dose < 2 mg bid during the 12-week double-blind randomised, placebo-controlled phase, were continued with their last dose from Period 1 regardless of treatment.
The primary objective of the study was to compare the proportion of complete responders (mUFC ≤ ULN) at the end of the 12-week placebo-controlled period between patients randomised to osilodrostat and the ones randomised to placebo. Patients who discontinued the study during the placebo-controlled period (n=2) or who had a missing mUFC assessment at Week 12 (n=3) were counted as non-responders for the primary endpoint. The key secondary objective was to assess the proportion of complete responders in both arms combined at Week 36 (mUFC ≤ ULN) in patients receiving osilodrostat. Dose reductions and temporary dose interruptions for safety reasons do not preclude patients from being counted as a complete responder for the key secondary endpoint.
Results.
In study C2302 the primary efficacy endpoint (proportion of complete responders at the end of the 12-weeks placebo-controlled period) was met. See Table 4.
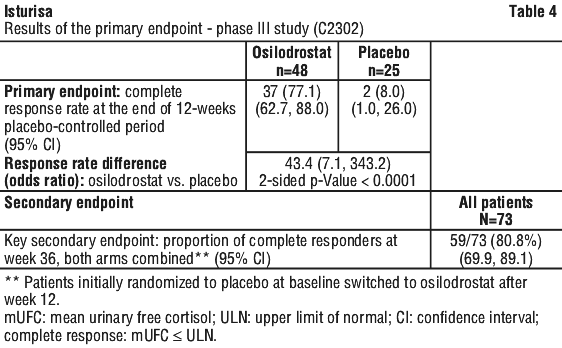 Overall, mUFC consistently decreased during treatment with osilodrostat. Median mUFC was reduced from 342.2 nanomol/24 h (2.5 x ULN) at baseline to 49.2 nanomol/24 h (0.4 x ULN; change from baseline - 83.6%) at Week 12 in patients treated with osilodrostat, while placebo patients median mUFC went from 297.6 nanomol/24 h (2.2 x ULN) at baseline to 305.5 nanomol/24 h (2.2 x ULN; change from baseline +4.5%).
Overall, mUFC consistently decreased during treatment with osilodrostat. Median mUFC was reduced from 342.2 nanomol/24 h (2.5 x ULN) at baseline to 49.2 nanomol/24 h (0.4 x ULN; change from baseline - 83.6%) at Week 12 in patients treated with osilodrostat, while placebo patients median mUFC went from 297.6 nanomol/24 h (2.2 x ULN) at baseline to 305.5 nanomol/24 h (2.2 x ULN; change from baseline +4.5%).
Osilodrostat treatment showed an improvement in cardiovascular-related clinical and metabolic parameters (e.g. fasting glucose, systolic blood pressure (SBP), diastolic blood pressure (DBP), weight, and waist circumference) associated with CD. The improvement in these parameters was already observed at the end of the placebo-controlled period (week 12) and maintained during the open-label treatment period (Week 12 to 48).
During the placebo-controlled period, there was a trend for more patients in the osilodrostat arm experiencing improvement in their physical features of CD, relative to the placebo arm. The exceptions were in the domains of facial rubor, striae, and proximal muscle atrophy.
Other causes of Cushing's syndrome. The efficacy of osilodrostat was also assessed in 9 adult Japanese patients with other causes of Cushing's syndrome (adrenal adenoma, ectopic corticotropin syndrome and ACTH-independent macronodular adrenal hyperplasia; study C1201). At Week 12 (primary endpoint), a complete response (mUFC ≤ ULN) was observed in 6 patients (66.7%) and a partial response (mUFC decrease by at least 50%) in one additional patient (11.1%). The median average dose used in the study was 2.6 mg/day (range 1.3-7.5 mg/day). The mean duration of treatment in this study was 24 weeks, and long-term exposure was limited.
5.2 Pharmacokinetic Properties
Absorption.
Osilodrostat is a highly soluble, highly permeable compound (BCS class 1). It is rapidly absorbed (tmax ~1 h) and oral absorption in humans is assumed to be nearly complete. Steady state is reached by day 2.
Co-administration with food did not affect absorption to a clinically significant extent. In a healthy volunteer study (n=20), the administration of a single dose of 30 mg osilodrostat with a high-fat meal resulted in a modest reduction of AUC and Cmax by 11% and 21%, respectively, and the median tmax was delayed from 1 to 2.5 hours.
No clinically relevant accumulation was observed in clinical studies. An accumulation ratio of 1.3 was estimated for the 2 to 30 mg dose range.
Distribution.
The median apparent volume of distribution (Vz/F) of osilodrostat is approximately 100 litres. Protein binding of osilodrostat and of its major metabolite M34.5 is low (less than 40%) and concentration-independent. The osilodrostat blood-to-plasma concentration ratio is 0.85.
Metabolism.
In a human ADME study in healthy subjects following the administration of a single dose of 50 mg [14C]-osilodrostat, metabolism was deemed the most important clearance pathway for osilodrostat since ~80% of the dose was excreted as metabolites. The three main metabolites in plasma (M34.5, M16.5 and M24.9) represented 51%, 9% and 7% of the dose, respectively. Both M34.5 and M24.9 have longer half-lives than osilodrostat and some accumulation is expected with twice-daily dosing. The decrease in the contribution of osilodrostat to the radioactivity AUC with time post-dose was found to coincide closely with a corresponding increase in the contribution of M34.5.
Thirteen metabolites were characterised in the urine, with the three main metabolites being M16.5, M22 (an M34.5 glucuronide) and M24.9, with 17, 13 and 11% of the dose, respectively. The formation of the major urinary metabolite M16.5 (direct N-glucuronide) was catalysed by UGT1A4, 2B7 and 2B10. Less than 1% of the dose was excreted as M34.5 (di-oxygenated osilodrostat) in the urine but 13% of the dose was identified as M22 (M34.5 glucuronide). The formation of M34.5 was non-CYP-mediated.
Multiple CYP enzymes and UDP glucuronosyltransferases contribute to osilodrostat metabolism and no single enzyme contributes more than 25% to the total clearance. The main CYP enzymes involved in osilodrostat metabolism are CYP3A4, 2B6 and 2D6. Total CYP contribution is 26%, total UGT contribution is 19% and non-CYP non-UGT mediated metabolism was shown to contribute to ~50% of total clearance. In addition, osilodrostat showed a high intrinsic permeability, low efflux ratio and modest impact of inhibitors on the efflux ratio in vitro. This suggests that the potential for clinical drug-drug interactions (DDI) with concomitantly administered medicinal products that inhibit transporters or a single CYP or UGT enzyme is low.
In vitro data indicate that the metabolites do not contribute to the pharmacological effect of osilodrostat.
Excretion.
The elimination half-life of osilodrostat is approximately 4 hours.
In an ADME study, the majority (91%) of the radioactive dose of osilodrostat was eliminated in the urine, with only a minor amount eliminated in the faeces (1.6% of dose). The low percentage of the dose eliminated in the urine as unchanged osilodrostat (5.2%) indicates that metabolism is the major clearance pathway in humans.
Linearity/non-linearity.
Exposure (AUCinf and Cmax) increased more than dose-proportionally over the therapeutic dose range.
Drug-drug interactions (also see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
In vitro data indicate that neither osilodrostat nor its major metabolite M34.5 inhibits the following enzymes and transporters at clinically relevant concentrations: CYP2A6, CYP2C8, CYP2C9, UGT2B7, P-gp, BCRP, BSEP, MRP2, OATP1B3 and MATE2-K. Since the exposure of M34.5 has not yet been determined after repeated dosing, the clinical relevance of the in vitro drug-drug interaction results for M34.5 is unknown.
Special populations.
Hepatic impairment.
In a phase I study in 33 subjects with varying degrees of hepatic function using a single dose of 30 mg osilodrostat, AUCinf was 1.4- and 2.7-fold higher in the moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment cohorts, respectively. Cmax was 15 and 20% lower in the moderate and severe cohorts. The terminal half-life increased to 9.3 hours and 19.5 hours in the moderate and severe cohorts. Mild hepatic impairment (Child-Pugh A) did not influence exposure to any significant extent. The absorption rate was not affected by the degree of hepatic impairment.
Renal impairment.
In a phase I study in 15 subjects with varying degrees of renal function using a single dose of 30 mg osilodrostat, comparable systemic exposure was seen in subjects with severe renal impairment, end-stage renal disease and normal renal function.
Race/ethnicity and bodyweight.
The relative bioavailability was approximately 20% higher in Asian patients compared to other ethnicities. Body weight was not shown to be a major determinant of this difference.
Age and gender.
Age and gender had no significant impact on osilodrostat exposure in adults. The number of elderly patients in clinical studies was limited (see Section 4.2 Dose and Method of Administration).
5.3 Preclinical Safety Data
Genotoxicity.
Osilodrostat was negative in the Ames assay and in the micronucleus test in mammalian cells, but clastogenic effects were observed in vitro at high concentrations in cultured human peripheral blood lymphocytes with and without a metabolic activation system. However, in vivo genotoxicity tests in rats (micronucleus assay and comet assay) were negative at sublethal doses. The weight of evidence from these studies suggests that osilodrostat does not pose a relevant genotoxic risk in humans.
Carcinogenicity.
In conventional 2 year rat and mouse carcinogenicity studies an increase in liver adenomas and/or carcinoma was seen in male mice dosed orally at 10 mg/kg/day (6 times the human clinical exposure at the MRHD) and at high doses in rats (10 mg/kg/day and 30 mg/kg/day in males and females, respectively; 23 and 74 times the human clinical exposure at the MRHD). Thyroid follicular cell adenomas and/or combined adenoma and carcinoma were also seen in male rats. The findings are likely rodent specific and considered not relevant to humans.6 Pharmaceutical Particulars
6.1 List of Excipients
Tablet core.
Microcrystalline cellulose, mannitol, croscarmellose sodium, magnesium stearate, colloidal anhydrous silica.
Film coat.
Hypromellose, titanium dioxide (E171), macrogol, purified talc, iron oxide yellow (E172) (1 mg, 5 mg and 10 mg tablet), iron oxide red (E172) (1 mg and 10 mg tablet), iron oxide black (E172) (10 mg tablet).
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C. Store in the original package in order to protect from moisture.
6.5 Nature and Contents of Container
Alu/Alu blisters with PA/AL/PVC foil as forming foil and aluminium foil as backing foil. Packs containing 60 tablets (6 blisters of 10 tablets).
6.6 Special Precautions for Disposal
No special requirements.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.

CAS number.
1315449-72-9.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription only medicine.
Summary Table of Changes
