


What is in this leaflet
This leaflet answers some common questions about Jakavi.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine. You can also download the most up to date leaflet from www.novartis.com.au. Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will provide.
If you have any concerns about this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Jakavi is used for
Jakavi is used to treat adult patients with myelofibrosis, a rare form of blood disorder with a variety of symptoms such as fever, night sweats, bone pain, weight loss. Enlarged spleen is one of the characteristics of myelofibrosis.
Jakavi is also used to treat patients with polycythemia vera who are intolerant of or not controlled with hydroxyurea. Polycythemia vera is a rare serious blood disorder with a variety of troublesome symptoms such as itching (pruritus), headache, vision problems, severe burning pain in the hands or feet, and blood vessels clots. Enlarged spleen is also sometimes present in patients with polycythemia vera.
Jakavi contains the active substance ruxolitinib phosphate.
This medicine belongs to a group of medicines called Jak inhibitors.
Myelofibrosis is a disorder of the bone marrow, in which the marrow is replaced by scar tissue. The abnormal marrow can no longer produce enough normal blood cells and results in a significantly enlarged spleen. Jakavi can reduce spleen size in patients with different forms of myelofibrosis and relieve the symptoms.
Polycythemia vera is a disorder of the bone marrow, in which the marrow produces too many red blood cells. The blood becomes thicker as a result of the increased red blood cells. Jakavi can relieve the symptoms, reduce spleen size and the volume of red blood cells produced in patients with polycythemia vera.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
Jakavi is not addictive.
Jakavi is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children under the age of 18 years.
Before you take Jakavi
When you must not take it
Do not take Jakavi if you have an allergy to:
- ruxolitinib, the active ingredient in Jakavi
- any of the other ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
Do not take this medicine if you are pregnant. It may affect your developing baby if you take it during pregnancy.
Do not breast-feed if you are taking this medicine. It is not known if the active ingredient in Jakavi passes into breast milk. There is a possibility that your baby may be affected.
Do not give this medicine to a child under the age of 18 years. There is not enough information to recommend the use of this medicine for children under the age of 18 years.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had (if appropriate) any of the following medical conditions:
- any infection
- problems with your kidney
- problems with your liver
- skin cancer
- tuberculosis
- viral hepatitis B
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
Tell your doctor if you have an intolerance to lactose. This medicine contains lactose.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Jakavi may interfere with each other. These include:
- some medicines used to treat fungal infections. These include medicines like ketoconazole, itraconazole, posaconazole and voriconazole
- some medicines used to treat types of bacterial infections. These include antibiotics like ciprofloxacin, clarithromycin, erythromycin or telithromycin
- some medicines used to treat viral infections
- some medicines used to treat fungal infections such as ketoconazole, itraconazole, posaconazole, fluconazole and voriconazole
- medicines used to treat HIV/AIDS such as atazanavir, indinavir, nelfinavir, ritonavir, saquinavir
- some medicines used to treat high blood pressure such as verapamil, nifedipine or carvedilol.
You may need different amounts of your medicines, or you may need to take different medicines. Your doctor and pharmacist have more information.
If you have not told your doctor about any of these things, tell him/her before you start taking this medicine.
How to take Jakavi
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you the dose that you should take. To find and maintain a suitable Jakavi dose for you, your doctor will check your blood cells and the condition of your liver and kidney. Your doctor may also need to know if you are on treatment with other medicines.
If you notice you are feeling unwell while taking Jakavi, your doctor might need to change the amount of Jakavi you have to take or tell you to stop taking Jakavi for a while.
Do not stop taking Jakavi unless your doctor tells you to.
How to take it
Jakavi tablets are to be taken by mouth, either with or without food.
Swallow the tablets whole with a glass of water.
When to take it
Take Jakavi twice a day, every day, at about the same time each day. If you receive dialysis, take one single dose of Jakavi before and another single dose following the dialysis. Your doctor will tell you how much the single dose is you need to take before and after the dialysis.
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How long to take it
Continue taking Jakavi for as long as your doctor tells you to.
This is long-term treatment. Your doctor will regularly monitor your condition to make sure that the treatment is having the desired effect.
If you interrupt your treatment with Jakavi, your myelofibrosis related symptoms may come back.
If you have questions about how long to take Jakavi, talk to your doctor or pharmacist.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take the next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking it as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Jakavi. Do this even if there are no signs of discomfort or poisoning. Keep the telephone numbers for these places handy.
While you are taking Jakavi
Things you must do
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some tests to check your blood cells and the condition of your liver and kidney from time to time to make sure the medicine is working and to prevent unwanted side effects. Your doctor may also regularly check the level of lipids (fat) in your blood.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking this medicine.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
Things you must not do
Do not give this medicine to anyone else, even if their condition seems similar to yours.
Do not use it to treat any other complaints unless your doctor tells you to.
Do not stop taking your medicine or lower the dosage without checking with your doctor. If you stop taking it suddenly, your condition may worsen or you may have unwanted side effects. If possible, your doctor will gradually reduce the amount you take each day before stopping the medicine completely.
Things to be careful of
Be careful driving, operating machinery or doing jobs that require you to be alert until you know how Jakavi affects you.
If you feel light-headed, dizzy or faint when getting out of bed or standing up, get up slowly. Standing up slowly, especially when you get up from bed or chairs, will help your body get used to the change in position and blood pressure. If this problem continues or gets worse, talk to your doctor.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Jakavi even if you do not think it is connected with the medicine.
All medicines can have side effects. Sometimes they are serious, but most of the time they are not. You may need medical treatment if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them. Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor as soon as possible if you notice any of the following:
- Urinary tract infection or any other infections
- Shingles (herpes zoster) - painful skin rash with blisters
- Bruising more easily than normal
- Fever, cough, difficult or painful breathing, wheezing, pain in chest when breathing (possible symptoms of pneumonia)
- Persistent cough with blood-tinged sputum, fever, night sweats and weight loss (possible symptoms of tuberculosis)
- Skin changes - may require further observation, as certain types of skin cancer (non-melanoma) have been reported.
The above list includes serious side effects which may require medical attention.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- Dizziness
- Headache
- Weight gain
- Excess amount of gas in the bowels (flatulence)
- Constipation
- Feeling more tired than usual
- Pale skin
- Frequent infections, fever, chills, sore throat or mouth ulcers due to infections.
The above list includes the more common side effects of your medicine. Most of these side effects are mild to moderate and will generally disappear after a few days to a few weeks of treatment.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Some people may have other side effects not yet known or mentioned in this leaflet. Other side effects can only be found when your doctor does tests from time to time to check your progress. These include:
- low level of red blood cells (anaemia), low level of white blood cells (neutropenia), low level of platelet (thrombocytopenia)
- high level of cholesterol (hypercholesterolemia) or fat in the blood (hypertriglyceridemia)
- abnormal liver function test results.
After using Jakavi
Storage
- Keep your medicine in the original container until it is time to take it.
- Store it in a cool, dry place at room temperature.
- Do not store this medicine or any other medicine in the bathroom or near a sink
- Do not leave it in the car or on window sills.
Keep the medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine you have left over.
Product description
What it looks like
Jakavi tablets are available in four different strengths, supplied in packs of 56 tablets.
- 5 mg: round curved and white to almost white tablets with "L5" on one side and "NVR" on the other
- 10mg: Round curved white to almost white tablets with "L10" on one side and "NVR" on the other
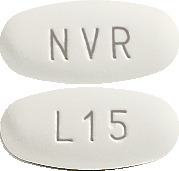
- 15 mg: ovaloid curved and white to almost white tablets with "L15" on one side and "NVR" on the other
- 20 mg: elongated curved white to almost white tablets with "L20" on one side and "NVR" on the other.
Ingredients
Jakavi contains 5, 10, 15 or 20 mg of ruxolitinib phosphate as the active ingredient. It also contains:
- microcrystalline cellulose
- magnesium stearate
- colloidal anhydrous silica
- sodium starch glycollate type A
- hyprolose
- povidone
- lactose monohydrate.
This medicine contains sugars in the form of lactose monohydrate. It does not contain sucrose, gluten, tartrazine or any other azo dyes.
Sponsor
Jakavi is supplied in Australia by:
NOVARTIS Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
Macquarie Park NSW 2113
Telephone 1 800 671 203
Web site: www.novartis.com.au
® = Registered Trademark
This leaflet was prepared in September 2021.
Australian Registration Number.
Jakavi 5 mg tablets AUST R 198934
Jakavi 10 mg tablets AUST R 232702
Jakavi 15 mg tablets AUST R 198936
Jakavi 20 mg tablets AUST R 198933
(jak210921c.doc) based on PI (jak210921i.doc)
Published by MIMS November 2021


 In GvHD, tapering of Jakavi may be considered in patients with a response and after having discontinued corticosteroids. A 50% dose reduction of Jakavi every two months is recommended. If signs or symptoms of GvHD reoccur during or after the taper of Jakavi, re-escalation of treatment should be considered.
In GvHD, tapering of Jakavi may be considered in patients with a response and after having discontinued corticosteroids. A 50% dose reduction of Jakavi every two months is recommended. If signs or symptoms of GvHD reoccur during or after the taper of Jakavi, re-escalation of treatment should be considered.


 Upon discontinuation MF patients may experience a return of myelofibrosis symptoms such as fatigue, bone pain, fever, pruritus, night sweats, symptomatic splenomegaly and weight loss. In MF clinical studies the total symptom score for myelofibrosis symptoms gradually returned to baseline values within 7 days after dose discontinuation.
Upon discontinuation MF patients may experience a return of myelofibrosis symptoms such as fatigue, bone pain, fever, pruritus, night sweats, symptomatic splenomegaly and weight loss. In MF clinical studies the total symptom score for myelofibrosis symptoms gradually returned to baseline values within 7 days after dose discontinuation.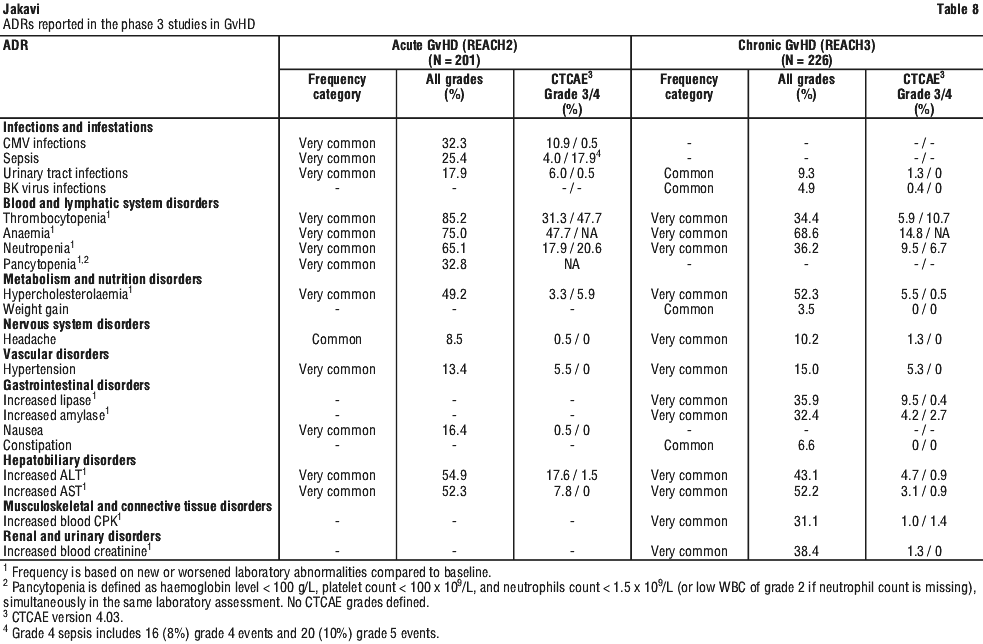
 In COMFORT-I, 41.9% of patients in the Jakavi arm achieved a ≥ 35% reduction in spleen volume from baseline compared with 0.7% in the placebo arm at Week 24. A similar proportion of patients in the Jakavi arm achieved a ≥ 50% reduction in the exploratory efficacy endpoint of palpable spleen length.
In COMFORT-I, 41.9% of patients in the Jakavi arm achieved a ≥ 35% reduction in spleen volume from baseline compared with 0.7% in the placebo arm at Week 24. A similar proportion of patients in the Jakavi arm achieved a ≥ 50% reduction in the exploratory efficacy endpoint of palpable spleen length. Figure 2 shows a waterfall plot of the percent change from baseline in spleen volume at Week 48 in COMFORT-II. Among the 98 patients in the Jakavi arm who had both baseline and Week 48 spleen volume evaluations, the median reduction in spleen volume at Week 48 was 28%. Among the 34 patients in the BAT arm who had both baseline and Week 48 spleen volume evaluations, there was a median increase of 8.5%.
Figure 2 shows a waterfall plot of the percent change from baseline in spleen volume at Week 48 in COMFORT-II. Among the 98 patients in the Jakavi arm who had both baseline and Week 48 spleen volume evaluations, the median reduction in spleen volume at Week 48 was 28%. Among the 34 patients in the BAT arm who had both baseline and Week 48 spleen volume evaluations, there was a median increase of 8.5%. The probability of duration from 1st ≥ 35% reduction of spleen volume to 25% increase from nadir and loss of response in COMFORT-I and COMFORT-II is shown in Table 10.
The probability of duration from 1st ≥ 35% reduction of spleen volume to 25% increase from nadir and loss of response in COMFORT-I and COMFORT-II is shown in Table 10. Among the 80 patients that showed a ≥ 35% reduction at any time point in COMFORT-I and of the 69 patients in COMFORT-II, the probability that a patient would maintain a response to Jakavi for at least 24 weeks was 89% and 87% in COMFORT-I and COMFORT-II respectively and the probability of maintaining a response for at least 48 weeks was 52% in COMFORT-II.
Among the 80 patients that showed a ≥ 35% reduction at any time point in COMFORT-I and of the 69 patients in COMFORT-II, the probability that a patient would maintain a response to Jakavi for at least 24 weeks was 89% and 87% in COMFORT-I and COMFORT-II respectively and the probability of maintaining a response for at least 48 weeks was 52% in COMFORT-II. Best overall response (BOR) is defined as the proportion of patients who achieved ORR (CR+PR) at any time point up to Cycle 7 Day 1. The BOR up to Cycle 7 Day 1 was higher in the Jakavi arm (76.4%) than in the BAT arm (60.4%).
Best overall response (BOR) is defined as the proportion of patients who achieved ORR (CR+PR) at any time point up to Cycle 7 Day 1. The BOR up to Cycle 7 Day 1 was higher in the Jakavi arm (76.4%) than in the BAT arm (60.4%).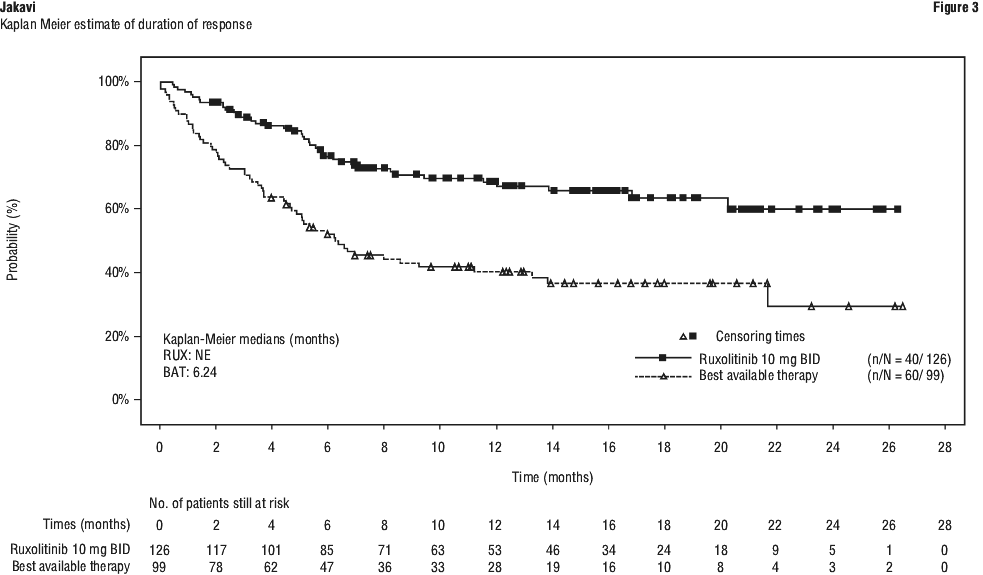
 The active ingredient of Jakavi is ruxolitinib (as the phosphate salt) or (R)-3-(4-(7H-Pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate.
The active ingredient of Jakavi is ruxolitinib (as the phosphate salt) or (R)-3-(4-(7H-Pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate.